Article Text

Abstract
Objective To describe the natural history of cardiomyopathy in patients with Duchenne muscular dystrophy (DMD) who are receiving contemporary therapies.
Methods This is a single-institution retrospective cohort study of 57 patients aged >15 years with DMD. Serial digital echocardiograms were performed over a median follow-up of 8 years. Left ventricular dysfunction (LVD) was defined as shortening fraction (SF) <29% plus focal wall motion abnormalities. Therapies included ACE inhibitors, beta-blockers and assisted ventilation.
Results The SF declined progressively in 53/57 patients (93%). LVD occurred in 40 of 57 patients (70%), with variable age at onset (median 18 years, IQR 14–21.5 years). Rate of SF decline (–1.51%±1.16%/year) was variable and unrelated to genotype. However, survival was shorter for patients with LVD onset at age <18 years vs onset at ≥18 years (death at 21.1±2.5 years vs 33.1±4.4 years; P<0.001). Death occurred in 27/57 (47%) patients at a median age of 26.3 years (IQR 20.6–31.5). Death was preceded by LVD in 22/27 patients (81%), 15 (68%) of whom developed class 4 heart failure (CHF). Time from CHF to death was brief (median 8.0 months).
Conclusion Despite contemporary therapies, SF declined progressively in almost all patients. Age at onset of LVD and age at death were variable and unrelated to genotype; however, survival was shortened for patients with LVD onset at age <18 years. Death was usually preceded by LVD. CHF was a sentinel event, with death occurring shortly thereafter.
- duchenne muscular dystrophy
- cardiomyopathy
- dystrophin genotype
- echocardiography
- heart function
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Key questions
What is already known about this subject?
Cardiac dysfunction is now a major cause of death in patients with Duchenne muscular dystrophy (DMD), and that the dystrophin genotype probably does not predict the course of the cardiomyopathy.
What does this study add?
Our long-term observations describe a progressive course of left ventricular dysfunction (LVD) despite treatment with contemporary cardiopulmonary therapies and show that LVD is highly variable in age of onset and unlikely related to genotype. We confirm that most deaths are of cardiac origin. A new observation is that onset of LVD at age <18 years is a predictor of early death. We document for the first time that survival after onset of class 4 heart failure is less than 1 year, making this a sentinel event.
How might this impact on clinical practice?
A better understanding of the contemporary natural history of DMD is needed to predict patient prognosis. Our demonstration of marked interpatient variability in the natural history of DMD cardiomyopathy and the lack of reliable genotype–phenotype correlations means that the efficacy of new therapies will be difficult to assess with aggregate data and is best judged in the context of each individual patient’s cardiac phenotype. For therapies to extend patient survival, they will need to delay the onset of LVD past 18 years of age and substantially postpone the onset of class 4 heart failure.
Introduction
Duchenne muscular dystrophy (DMD) is caused by an X linked mutation within the dystrophin gene at the Xp21.1 locus1 and is characterised by degeneration of skeletal and cardiac muscle, leading to loss of ambulation, respiratory insufficiency and progressive cardiac dysfunction.2 As respiratory insufficiency is now well managed, cardiac dysfunction has become a common cause of death.3–5 However, the contemporary natural history of cardiac dysfunction in DMD has not been described and cardiac prognostic predictors are unknown. For example, age of onset of left ventricular dysfunction (LVD) does not reliably correlate with either skeletal muscle weakness or pulmonary function abnormalities,6 and the correlation between dystrophin genotypes and cardiac phenotypes remains uncertain.7–10 Current DMD guidelines recommend close surveillance for LVD and, if present, initiation of ACE inhibitor (ACEI) therapy.11 However, it is unknown if this approach consistently alters the natural history of DMD cardiomyopathy. Herein, we describe the modified natural history of cardiac function in patients with DMD receiving contemporary cardiopulmonary therapies and seek clinical variables that predict cardiac course.
Materials and methods
Study design and participants
This is a retrospective cohort study of patients with DMD followed at MetroHealth Medical Center Muscular Dystrophy Association Clinic. The study’s inclusion criteria were a diagnosis of DMD based on typical clinical characteristics, a markedly elevated serum creatine kinase level, a mutation of the dystrophin gene consistent with DMD and ≥3 readable echocardiograms from the time frame 2003 through 2015. Exclusion criteria were patients aged <16 years at the time of their last echocardiogram (due to low likelihood of LVD and clinical heart failure in these young patients), <3 readable echocardiograms from the eligible time frame or inadequate patient follow-up (figure 1).

Flow chart of patient cohort.
Assessment of left ventricular function
Echocardiograms were obtained annually whenever possible as part of routine care. Imaging was performed using either Philips Sonos 500 or Philips iE33 (Andover, Massachusetts) systems and stored in a digital format. All relevant echocardiographic parameters were remeasured by one of two authors (MW, RCB) who were blinded to clinical data. Concordance between authors was assessed by randomly selecting 15 echocardiograms for independent measurements. Some digital images (n=15) prior to 2005 were not retrievable so clinical report data were abstracted. Left ventricular (LV) dimensions were obtained from parasternal long axis views. The LV minor axis dimension at end-diastole (LVIDd) was measured at the mitral chordae level or at the first frame of isometric contraction if gastric compression of the inferolateral wall reduced the minor axis dimension. End-systolic dimension (LVIDs) was measured at the same location at the smallest systolic diameter. Shortening fraction (SF) was calculated as {[(LVIDd–LVIDs)/LVIDd] ×100}. The SF incorporates the basilar aspect of the inferolateral wall, the predominant area of early LV dysfunction in DMD. All standard LV views were reviewed for evidence of abnormal wall motion. The presence of both SF <29% and focal wall motion abnormalities defined LVD. Mitral inflow velocities were obtained from the apical four-chamber view. The SF rather than LV ejection fraction was selected to characterise changes in LV function since biplane apical images, required for reliable measurement of LV ejection fraction, become problematic in older patients with DMD as loss of acoustic windows follows loss of intercostal muscle. Difficulty of peripheral venous access precluded use of intravenous contrast. The echocardiographic measurement of SF in patients with DMD reasonably reflects the LV ejection fraction as assessed by cardiac magnetic resonance imaging (CMR).12
Patient information
The electronic medical record was reviewed for medications including beta-blockers, ACEI, angiotensin receptor blockers (ARB), digoxin, steroids and diuretics. Therapies were initiated after the onset of LVD in 45 patients and before LVD onset in 12 patients. Class 4 heart failure (CHF) was defined by dyspnoea, orthopnoea, fluid retention and the initiation of diuretic therapy. Supporting data included radiological evidence of pulmonary congestion and an elevated B-type natriuretic peptide. Date of death was obtained from the State of Ohio Bureau of Vital Statistics.
Genotyping
Mutations within the dystrophin gene were identified using one or more of the following methods: PCR, southern blots, DMD gene sequencing and/or genomic hybridisation array, depending on the technology that was available at the time of diagnosis.9
Statistical analysis
Data are reported as follows: means with the corresponding SD for parametric, interval data; medians with the IQR for non-parametric interval or ordinal data; and percentages for categorical data. Parametric, interval data are defined as having both a value for skewness and kurtosis between −3 and +3. Differences between groups were analysed using the following statistical tools: Χ2 or Fisher’s exact test for categorical data; Mann-Whitney U test for ordinal data or non-parametric interval data; and t-test for parametric interval data. Log-rank test was used in comparing the survival curves if the assumption of proportional hazard was met. If the assumption was not met, then Tarone-Ware test was used. Concordance between the two echocardiogram readers was assessed using intraclass correlation coefficient. Differences among the three genotypes were analysed using Kruskal-Wallis test. If the P value was <0.05, then the appropriate post-hoc comparisons were performed using Mann-Whitney U test coupled with Bonferroni correction. Data were analysed using IBM SPSS Statistics V.23. Statistical significance was defined a priori as a ‘p value’ <0.05, two-tailed.
Results
Patient characteristics
Review of the patient registry identified 103 patients with dystrophin gene mutations, 93 of whom had a clinical diagnosis of DMD. Applying inclusion/exclusion criteria left a study cohort of 57 patients (figure 1). Patients’ clinical characteristics are shown in table 1.
Patient characteristics
The mean age at the time of the first and last echocardiograms was similar for patients with and without LVD. Patients were followed for a median of 8 years. Over 97% of patients with LVD received ACEI or ARB therapy, and two-thirds of patients with LVD were treated with a beta-blocker. Patients with CHF were treated with diuretics. Long-term steroid therapy was uncommon (10 patients). All but seven patients received non-invasive ventilation or ventilation via tracheostomy.
Echocardiographic observations
The number of echocardiograms reviewed per patient ranged from 3 to 14 (mean 7.2±2.9). Echocardiographic measurements were reproducible with interobserver variability, expressed as intraclass correlations, as follows: LVIDd r=0.98, LVIDs r=0.97 and SF r=0.82.
A progressive decline in SF occurred in 53 of 57 patients. Their rate of decline was −1.51±1.16% per year. Forty (70.2%) patients exhibited LVD during their clinical course. Age at onset of LVD varied markedly with a median value of 18 years (IQR 14–21.5 years). All patients with LVD demonstrated a decline in SF despite contemporary therapies (table 2).
Echocardiographic data comparing those with versus without LV dysfunction during follow-up
Marked individual variability occurred in the % change in SF per year (range: −0.45% to −3.8% per year). For patients with LVD, the LVIDd was significantly larger in the final echocardiogram (table 2). The difference between the LVIDd in the last versus the first echocardiogram was even greater among patients with CHF (with initial and final values of 5.1±0.9 cm and 6.8±0.6 cm, respectively; P<0.001).
Dystrophin genotypes and cardiac phenotype
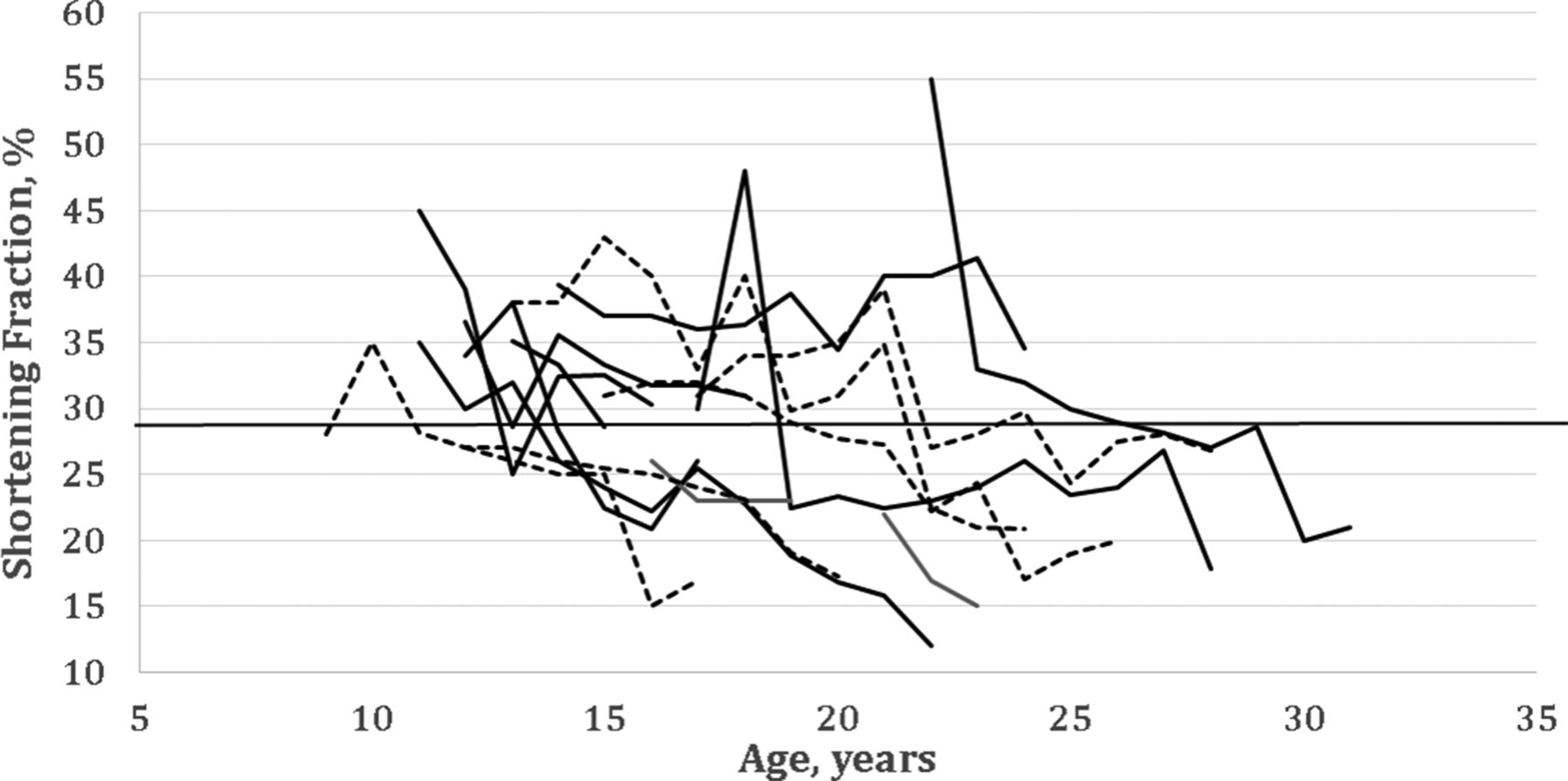
We assessed the cardiac course of three genetic groups: group A, isolated deletion of exon 44, n=9; group B, isolated deletion of exon 51, n=5; group C, a diverse comparison group of the remaining patients with different mutations, each mutation shared by less than three patients (n=43). Cardiac dysfunction was unpredictable and highly variable both among the individual patients with identical mutations (groups A and B; figure 2) and in the intergroup comparisons of cardiac function (table 3).

Plots of shortening fraction (SF) versus age for patients with identical genotypes. Patients with deletion of exon 44 (solid lines) or deletion of exon 51 (dotted lines) exhibit variability in the age when the SF falls below 29% (solid horizontal line) even when having identical genotypes. Subsequently, the SF continues to decline at similar rates.
LV dysfunction and genotypes
The individual ages of onset of LVD were strikingly variable (group A range: 14–29 years, group B: 10–22 years; and group C: 10–40 years). There was no apparent association between a genetic group and onset of LVD at age <18 years as compared with onset at ≥18 years.
All genetic groups demonstrated a decline of SF over time (described by calculating the regression slope of SF versus age for each patient and then determining the mean slope for each group), and this decline did not differ significantly between groups (table 3). All three genetic groups demonstrated an increase in LVIDd over time (table 3).
Patient outcomes
The significant contributions of LVD and CHF to outcomes are shown in table 4.
Cardiovascular outcomes
Causes of death for the seven patients with LVD but no CHF included sepsis (1), pulmonary infections (3), other respiratory complications (2) and unknown or ‘sudden’ (2). Of the 40 patients who exhibited LVD at some point during the observation period, 15 (38%) progressed to CHF at a mean age of 25±6.9 years.
Survival analyses indicated that the cumulative survival of group A patients over time was similar to the cumulative survival of patients in groups B and C combined (figure 3A). Death occurred at a significantly earlier age for patients with onset of LVD at age <18 years compared with patients with LVD onset at age ≥18 years (age at death 21.1±2.5 years vs 33.1±4.4 years, respectively; P<0.001; figure 3B). Although the mean age at death was significantly different for the two groups (P<0.001), the rate of death per time appeared similar. Furthermore, the slope of decline in SF did not differ between those with onset of LVD at age <18 compared with those with onset at age ≥18 years (−1.61±0.88 vs −1.20±0.95, respectively; P=0.166). Patients developing CHF had a reduced survival compared with those with LVD but no CHF (P=0.005; figure 3C). Onset of CHF was a sentinel event, with a median survival from the onset of CHF to death of just 8 months (IQR 2.5–19 months, n=15 patients) (figure 3D).

{kind=link}
{kind=link}
{kind=link}
(A) Survival for those with deletion of exon 44 (n=9) versus the remaining cohort (n=48). The survival curves are similar, although the duration of follow up was less for those with a deletion of exon 44. The median survival time for the entire cohort was 31.7 years (95% CI 27.4 to 36.0 years). (B) Survival of patients with onset of LVD at age <18 years (n=18) vs age ≥18 years (n=22). Death occurred at an earlier age if LVD developed at age <18 years (P<0.001). The slopes of the survival curves appeared similar. (C) Survival after onset of LVD in patients with (n=15) and without (n=25) CHF. The development of CHF reduced the survival time (P=0.005). (D) Survival after onset of CHF (n=15). Survival was brief after onset of CHF. CHF, class 4 heart failure; LVD, left ventricular dysfunction.
Discussion
Despite the use of contemporary cardiopulmonary therapies, all patients observed more than 5 years experienced a progressive decline in heart function which progressed to LVD in most. However, both the rate of decline in cardiac function and the age at onset of LVD were variable. Once LVD advanced to CHF, survival was brief, with death usually occurring within 1 year. A cardiac phenotype was identified in that patients with onset of LVD at age <18 years died at a younger age than those with onset of LVD at ≥18 years, suggesting that early onset of LVD may be a good marker for patients with a more severe cardiac phenotype.
These results imply that contemporary therapies did not significantly alter the relentless progression of heart dysfunction; that is, therapies did not achieve significant reverse remodelling of DMD cardiomyopathy. If our results are replicated, then the inability of contemporary treatments to alter the long-term progression of DMD cardiomyopathy is distinct and contrasts with that seen in idiopathic cardiomyopathy, where therapy can alter the progression of LV dysfunction and reverse remodelling is common.13 14 It is not surprising that the DMD cardiomyopathy is progressive since ongoing myocyte death is consequent to the genetic abnormality. As LV dilatation ensues, the increased wall stress aggravates the progression of LVD.
Short-term outcome data
Short-term studies in predominantly younger patients have reported that medications improve LV function in DMD cardiomyopathy.15–17 However short-term observations could be confounded by random variations in LV ejection fraction or by the chance assignment of patients with a favourable cardiac phenotype to the active therapy group. In contrast, our findings of progressive LVD are supported by a recent study using the more sensitive diagnostic modality of CMR, which showed a decline in LV function and LV myocardial strain in a young cohort despite ongoing therapy.18 The view that ACEI/ARB therapy has limited efficacy over time is also supported by a recent analysis of these therapies in adults with LV systolic dysfunction of diverse aetiologies.19
Other outcome data
Long-term longitudinal outcome data for the DMD cardiomyopathy are sparse. Several studies suggest that our results may be representative of DMD populations in general. An echocardiographic study found the mean age at onset of cardiomyopathy was 16.5 years (range 3.6–36.5 years), comparable with our finding of LVD onset at a median age of 18 years.20 A 1-year echocardiographic study in patients undergoing surgery for scoliosis showed the SF percentage decreased by approximately 1% per year,21 like our cohort in whom SF declined at a mean rate of 1.51±1.16% per year. The multicentre Pediatric Cardiomyopathy Registry report included 128 patients with DMD from 57 centres. These retrospective observations were prior to 1995; only a small minority received an ACEI or ARB, none received a beta-blocker and it is unclear what proportion received home respiratory support. Death occurred in 50% of patients within 5.5 years of the diagnosis of cardiomyopathy,22 comparable with our actively treated cohort with a median survival of 6.8 years after onset of LVD. A recent CMR study showed steady progression of LVD and myocardial fibrosis over several years, with the LV ejection fraction declining at a rate of 0.58%±0.10% per year, more rapidly if late gadolinium enhancement was evident.23 Their young cohort had few outcome events. Cardiac function was assessed in a young cohort using CMR; LV ejection fractions tended to decrease and LV global circumferential strain showed a significant decline despite 1 year of ACEI/ARB treatment.18 These data support our finding that once LVD is evident, there is usually steady progression of cardiac dysfunction, suggesting current therapies do not substantially alter the natural history of DMD cardiomyopathy.
CHF and death
The extremely short survival of patients who developed CHF is a new and important observation; CHF is a sentinel event with dire prognostic implications. This observation contrasts with that of idiopathic dilated cardiomyopathy where treatment often reverses CHF symptoms and a sustained clinical improvement follows.13 We suggest that a therapy for DMD cardiomyopathy can only be considered effective if it prevents or delays onset of CHF. Onset of CHF could be used both as an outcome measure and as a negative prognostic marker. Our finding implies that patients with CHF are not good candidates for implanted intracardiac defibrillators since current guidelines include a ‘reasonable expectation of meaningful survival for more than 1 year’.24
Genotype–phenotype correlations, cardiac natural history and cardiac phenotypes
Our modest sample size precludes a definitive answer as to whether differences exist in outcomes between different dystrophin genotypes.25 However, our observation that patients with identical exon deletions have highly variable ages at onset of LVD implies that the clinical course is not predicted by the genotype. Additionally, classifying cardiac phenotypes by age at onset of LVD could have important prognostic implications: patients with onset of LVD at <18 years had a severe phenotype and experienced an earlier death. However, there was no clear association between a patient’s dystrophin genotype and the presence of either the severe or mild cardiac phenotype, suggesting a patient’s dystrophin genotype cannot be used to predict the trajectory of his cardiac function over time. This observation has important implications for the evaluation of current and future therapies; that is, efficacy of cardiac therapies should be evaluated in the context of each individual patient’s cardiac phenotype. For example, good heart function over time might be mistakenly attributed to a therapy when the cause is attributable to a favourable cardiac phenotype. Regarding the aetiology of cardiac phenotypic variability, it is likely that cardiac function is related not just to a patient’s dystrophin mutation, but to his entire genetic milieu, including the presence of detrimental or beneficial cardiac modifier genes.20
Study strengths and limitations
A single-institution observational study provides consistency in analysis of echocardiographic data, a standardised approach to therapies and a high level of detail regarding each patient’s clinical course. However, our retrospective cohort design means that we cannot determine causal relationships. The small size of our cohort means that our observations require confirmation to assure that they are generalisable.
The advantages and limitations of echocardiography as our primary imaging modality deserve discussion, since CMR has become the preferred diagnostic modality.23 25 Echocardiographic measures of cardiac function were chosen because CMR with late gadolinium enhancement for detection of fibrosis was not commonly available at the time of their presentation, and many older patients would have been unsuitable (ie, spinal rods and/or intolerance of the physical demands of CMR). Assessment of SF by echocardiography was shown to correlate with LV ejection fraction by CMR in cohorts of young patients with muscular dystrophy.12 26 Since CMR is a more sensitive diagnostic modality, it is probable that a greater proportion of our patients would have been identified as manifesting cardiac dysfunction and at an earlier age had we used CMR as our imaging modality.23 27 28 Overall, our observations regarding progression of LV dysfunction and patient outcomes are valid and independent of the imaging modality. Importantly, echocardiography remains the most widely used imaging modality for patients with DMD.
The retrospective design of our study meant we could not always ascertain the age at onset of LVD. Additionally, patients <16 years of age were evaluated by paediatric cardiologists from another institution and digital storage of the echocardiographic images was not performed. This precluded an analysis of serial LV function. Variations in medication dosage might confound the natural history of LVD, but our dosing regimens were similar for the patients with LVD. Additionally, the ATLAS trial of low-dose versus high-dose lisinopril did not find a marked difference in patient outcomes.29
Conclusion
Our longitudinal study of cardiac function in patients with DMD found that SF declined over time in most patients despite contemporary cardiopulmonary therapies and in all patients after onset of LVD. Once CHF appeared, death was imminent. Within this general pattern, there was marked interpatient variability in both the rate of progression and age at onset of LVD. The median age at death was 26.3 years, but when LVD occurred at <18 years of age survival was shortened. Dystrophin genotype did not predict the rate of SF decline, age at onset of LVD, or a mild or severe cardiac phenotype.
Acknowledgments
We thank Dr Douglas Einstadter for obtaining the dates of patient deaths.
References
Footnotes
Contributors All authors participated in the study design. MW and RCB were responsible for abstracting both the echocardiographic data and the data from the medical records. The statistical analysis was performed by DMS and RCB. All authors contributed to the revisions of the manuscript and approved the final version.
Funding This research received no specific grant from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests None declared.
Patient consent Not required.
Ethics approval The study was approved by the MetroHealth Medical Center Institutional Review Board (IRB# IRB06-00655).
Provenance and peer review Not commissioned; internally peer reviewed.
Data sharing statement There are no unpublished data apart from the content of the submitted manuscript.