Article Text

Abstract
Aims To define the prevalence of non-sustained tachyarrhythmias and bradyarrhythmias in patients with the m.3243A>G mitochondrial genotype and a previously defined, profile, associated with ‘high sudden-death risk’.
Methods and results Patients at high risk of sudden death because of combinations of ventricular hypertrophy, mitochondrial encephalopathy, lactic acidosis and stroke-like episodes family phenotype, epilepsy or high mutation load, due to the m.3243A>G mutation, were identified from a mitochondrial cohort of 209 patients. All recruited had serial ECG and echo assessments previously according to schedule, had an ECG-loop recorder implanted and were followed for as long as the device allowed. Devices were programmed to detect non-sustained brady- or tachy-arrhythmias. This provided comprehensive rhythm surveillance and automatic downloads of all detections to a monitoring station for cardiology interpretation. Those with sinus tachycardia were treated with beta-blockers and those with ventricular hypertrophy received a beta-blocker and ACE-inhibitor combination.
Nine consecutive patients, approached (37.2±3.9 years, seven males) and consented, were recruited. None died and no arrhythmias longer than 30s duration occurred during 3-year follow-up. Three patients reported palpitations but ECGs correlated with sinus rhythm. One manifest physiological, sinus pauses >3.5 s during sleep and another had one asymptomatic episode of non-sustained ventricular tachycardia.
Conclusions Despite ‘high-risk’ features for sudden death, those studied had negligible prevalence of arrhythmias over prolonged follow-up. By implication, the myocardium in this genotype is not primarily arrhythmogenic. Arrhythmias may not explain sudden death in patients without Wolff-Parkinson-White or abnormal atrioventricular conduction or, it must require a confluence of other, dynamic, proarrhythmic factors to trigger them.
- arrhythmias
- hypertrophic cardiomyopathy
- inborn genetic diseases
- genetics
Data availability statement
All data relevant to the study are included in the article or uploaded as online supplemental information.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Key questions
What is already known about this subject?
The frequency of sudden death in patients with the m.3243A>G mitochondrial genotype is estimated to be 2.4/1000 patient years. These deaths have been assumed due to ventricular tachycardia/fibrillation because mitochondrial disorders can cause left ventricular hypertrophy and some myocardial scaring. However, the cardiac phenotype is mild, with a scar burden unlikely to be sufficient to sustain arrhythmias on its own.
What does this study add?
The finding of a negligible burden of non-sustained arrhythmias during comprehensive, prolonged, ECG-rhythm surveillance argues against the myocardium being primarily arrhythmogenic in the m.3243A>G mitochondrial disorder.
How might this impact on clinical practice?
By implication, sudden deaths in patients may not be due to cardiac arrhythmias at all or, alternatively, arrhythmias may only be able to occur when metabolic disturbances or other proarrhythmic contributors transiently coincide. The mechanism of sudden death in the m.3243A>G mitochondrial disorder remains unexplained.
Introduction
Mitochondrial disorders are a heterogeneous group of multisystemic diseases that develop as a result of mutations in nuclear or mitochondrial DNA.1 These conditions are estimated to affect about 1 in 5000 births.2 3 Cardiac involvement varies but can include the development of left ventricular hypertrophy, systolic and/or diastolic left ventricular dysfunction with or without heart failure, Wolff-Parkinson-White (WPW)-syndrome, progressive atrioventricular (AV)-nodal conduction delay and sudden death (SD).4 5 In a retrospective analysis of 691 patients with various mitochondrial genotypes, under regular follow-up by the Newcastle based, National Health Service (NHS) Highly Specialised Service and Wellcome Centre for Mitochondrial Research, we demonstrated previously that cardiac involvement occurred in 25% of patients with the most common form of mitochondrial disease—that due to the m.3243A>G variant.4 SD in this genotype has not been studied systematically, despite reports of it occurring in patients who were not considered to be at arrhythmia risk because of having only a minor degree of left ventricular hypertrophy, no pre-excitation and no delay in AV conduction.6–9 By extrapolation from our large cohort of patients with complete follow-up, we estimated the incidence of SD in these patients to be 2.4/1000 patient years.9 Although little stratification of arrhythmia risk is undertaken routinely, we proposed a number of risk factors associated with SD in patients: maximum left ventricular wall thickness greater than 15 mm by echocardiography; family history of sudden cardiac death (SCD) or severe mitochondrial encephalopathy, lactic acidosis and stroke-like episodes (MELAS) phenotype; epilepsy; high blood m.3243A>G mutant load.9
SD could be due to diverse causes—ventricular tachy-arrhythmias (ventricular tachycardia/fibrillation), profound bradycardia/asystole, electromechanical dissociation (pulseless electrical disease) or to non-cardiac causes such as epilepsy-associated SD or major aspiration due to bulbar dysfunction. SCD could be facilitated by either fixed (eg, fibrosis, premature failure of the conduction system, WPW) or dynamic arrhythmia substrates (eg, triggered by metabolic derangement such as hypoglycaemia or lactic acidosis).10–12
This study aimed to determine whether the myocardium of patients with the m.3243A>G mitochondrial disorder is primarily arrhythmogenic by documenting the spontaneous arrhythmia ‘burden’ (ie, non-sustained, minimally symptomatic) over prolonged, comprehensive, ECG-rhythm surveillance.
Methods
Two hundred and nine patients with mitochondrial disease due to the m.3243A>G variant and who are under regular follow-up by the NHS Highly Specialised Service for Rare Mitochondrial Disorders in Newcastle upon Tyne were identified from pre-existing data sets. As part of routine clinical care, all had previously had their clinical status characterised comprehensively in terms of its clinical phenotype (ie, epilepsy; diabetes; stroke-like and encephalitis episodes; renal; other system involvement), disease burden as measured by Newcastle Mitochondrial Disease Adult Scale,13 and genetic mutation as well as mutant load in blood and urine. The results of serial ECGs and echocardiographic measures of heart function and wall thicknesses were also available. Together this data-set allowed patients with any of the five high risk features for SD, identified from our earlier retrospective analysis (ie, LVH >15 mm by echo-measurement; family history of SD; mutation load blood heteroplasmy > (79.6–15 × age); family history of MELAS phenotype; epilepsy), to be identified as eligible for inclusion. Some had data from cardiac MR-imaging, performed for clinical reasons, to determine the extent of cardiac involvement and presence intramyocardial scaring12
In advance of a routine follow-up clinic visit, those eligible for inclusion on the basis of mutation type were sent a patient information document, informing them of the study and what participation involved. Patients interested in taking part then had a discussion with a member of the clinical research team, in the course of which, they were made aware that their clinical and genetic profile carried a risk of SD Those agreeing to take part, gave written informed consent and were recruited to the study.
Participants then had an ECG loop-recorder implanted (Reveal or LINQ Insertable Cardiac Monitor, Medtronic, USA) subcutaneously in the left parasternal area in a day-case procedure under local anaesthesia.14 15 Devices were programmed to detect pathological brady (ie, heart rate <30 /min for >3 s) or tachycardia (ie, heart rate >150/ min for >3 beats) episodes automatically. Each patient was provided with a unique remote telemetry device, which allowed full disclosure of all predefined ECG-episodes detected, and these were transmitted automatically to a commercially provided website. All recordings were reviewed by a cardiologist at the implanting centre, along with their date, time, nature and a high-quality ECG of each episode. This was all possible without the patient having to re-attend the hospital. Patients were also shown how to make recordings at time of any symptoms, using a hand-held device-activator. Implantable loop-recorders have battery life of more than 3 years. The primary aim was to define the spontaneous ‘arrhythmia burden’ over prolonged follow-up (ie, until transmissions ended. battery expiry or censure 31 December 2019) as an indirect measure of the arrhythmogenicity of the myocardium in this condition. A secondary aim was to correlate arrhythmias with the occurrence of any illnesses (eg, seizures/encephalopathy; infection; hypoglycaemia) over the same time period.
Patient and public involvement
Neither patients nor the public were involved in the design, conduct, reporting or dissemination plans of this exploratory study of enhanced arrhythmia surveillance.4
Results
From a cohort of 209 patients with the m.3243A>G mitochondrial genotype, nine consecutively recruited patients, mean age at implant 37.2±3.9 years, seven males, were consented and had their device implanted in the period February–October 2016. Data collection was censured on 31 December 2019. Participant age categories and m.3243A>G heteroplasmy levels are summarised in table 1 and examples of ECG and cardiac MR-abnormalities in figures 1 and 2).

Examples of ECG abnormalities in the patients studied. P1,5,6 correspond to patient numbering in table 1; P1—left ventricular hypertrophy and ‘strain’ pattern; P5—pathological left axis QRS—deviation and left atrial hypertrophy; p6—repolarisation changes (T-wave ‘flattening’) V5-6. aVF, augmented vector foot; aVL, augmented vector left; aVR, augmented vector right.

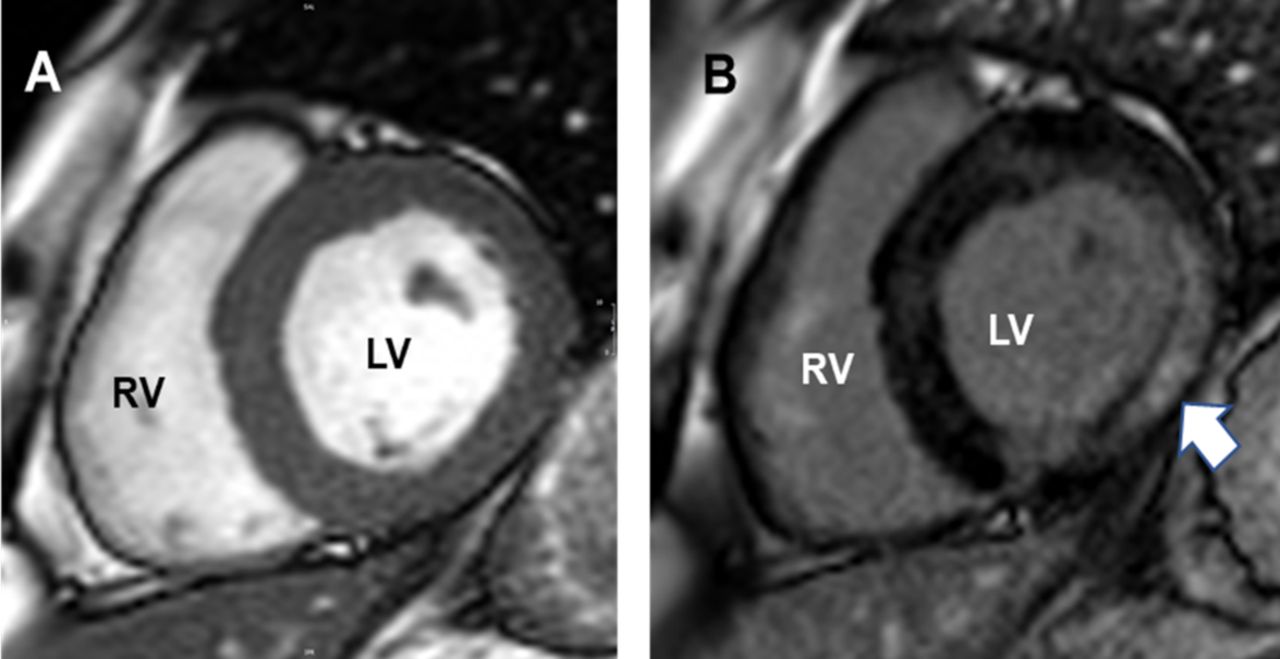
Cardiac MI images of patient with heart involvement in mitochondrial disease. Short axis, mid-ventricular cardiac MR image of left (LV) and right ventricles (RV) (A) of patient ‘5’. (B) shows extensive intramyocardial fibrosis (arrowed) in the lateral LV-wall on late-Gad sequences due to m.3243A>G mitochondrial disorder.
Patient age categories, m.3243A>G phenotype and cardiac findings
Their clinical features listed were all attributable to the m.3243A>G variant. Those recruited had a wide age range and were at varying stages in the evolution of their mitochondrial phenotype. All had genetic and some clinical features to indicate that they were at increased risk of SD.9
Patient and/or ECG-recorded events during follow-up
During 3 years of follow-up, three participants activated their devices on a total of 20 occasions to make recordings because of symptoms. None of the corresponding ECGs detected anything other than sinus rhythm at varying rates. Devices ‘captured’ 68 ECG-episodes automatically in nine patients. Sixty-four (94%) of these were ‘false positives’, meaning that the corresponding ECGs confirmed sinus tachycardia without ectopy or any pathological bradyarrhythmias (table 2).
Device activations (automatic or patient activated) during 3-year follow-up
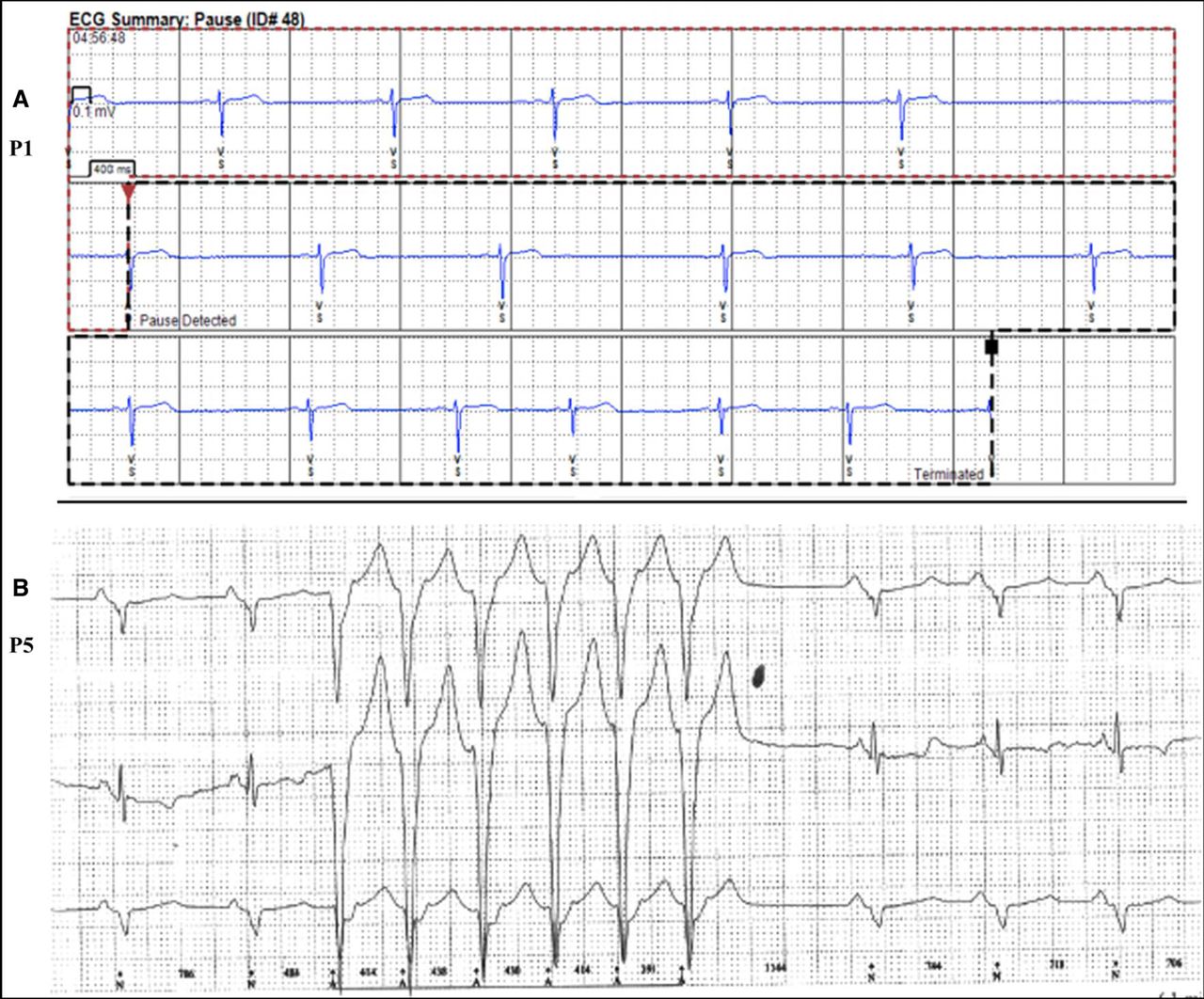
One patient, known to have sleep apnoea, had a single 3.6 s sinus pause, during sleep (figure 3A) and another (P5) had an asymptomatic run of non-sustained broad-complex tachycardia recorded on one occasion (figure 3B). The proportion of ‘false’ automatic, device activations is consistent with loop-recorders having higher sensitivity than specificity in arrhythmia detection.14 15

{kind=link}
{kind=link}
{kind=link}
(A and B) Arrhythmias recorded over 3-year surveillance. (A) Sinus bradycardia and 3 s sinus arrest during sleep—median ventricular rate 41 /min (B) Automatic recording of asymptomatic ‘run’ of irregular, non-sustained ventricular tachycardia. ECG recording speed 25 mm/s.
Effects of episodes of intercurrent illness on arrhythmia detections
Only two patients (P2&3) experienced significant episodes of illness over the 3 years when their devices were able to record and transmit ECGs. One had a series of close-coupled seizures and frequent ‘absences’ and the other gastroenteritis with vomiting and dehydration, requiring hospital admission. Neither manifested arrhythmia symptoms, nor were any arrhythmias detected automatically at these times.
Discussion
Previously, our group proposed ‘high-risk’ features associated with the occurrence of SD in patients with m.3243A>G-related mitochondrial disease and estimated the event rate to be 2.4/1000 patient years.9 The mechanisms underlying SD, however, have not been studied systematically. This is the first study which aimed to determine the burden of spontaneous arrhythmias, occurring over prolonged follow-up in a uniformly well characterised ‘high risk’ group, using implanted loop recorders as a measure of myocardial electrical instability in this disorder. There were no deaths, none developed sustained arrhythmias and only two showed any abnormality—one, known to have sleep apnoea, had a three-second sinus pause during sleep and another had a six-beat ‘run' of ventricular tachycardia on one occasion. Even in those reporting palpitations, symptoms correlated only with sinus rhythm at varying rates. The essentially negative findings argue against the myocardium being primarily arrhythmogenic in this mitochondrial disorder.
The prevailing assumption, that arrhythmias account for SD in the m.3243A>G-related disease, is based on evidence of arrhythmias occurring in patients with a quite different mitochondrial disorder—Kearns-Sayre syndrome (KSS).6 7 16–19 In KSS symptomatic bradycardia, asystole and SD have been documented to occur as a progression of, previously identified, AV-conduction abnormalities.7 16 Others describe cases of ventricular tachyarrhythmias—including torsades-de-points16 17 or ventricular fibrillation,18 occurring in the context of recurring episodes of bradycardia or complete AV-heart block. The importance of these reports is, first, in highlighting the need for surveillance to identify progressive abnormalities in AV-conduction in KSS and, second, the role of prophylactic pacing in preventing SD from either asystole or bradycardia-induced ventricular fibrillation in that particular form of mitochondrial disease.6 However, such patients may still be capable of sustained ventricular tachycardia, despite pacing, when damage to the intraventricular conduction system or the extent of myocardial fibrosis can support localised or bundle branch re-entrant arrhythmia circuits.19 This has led some to suggest that when patients with KSS come to device implant, they should receive an implantable cardioverter defibrillator rather than a simple pacing device.19 Taken together, these reports all implicate fibrotic damage to the AV-conduction system as the primary contributor to arrhythmias in the KSS form of mitochondrial disorder. There are similarities between some of the mechanisms underlying life-threatening arrhythmias in KSS and those that contribute to arrhythmias in hypertrophic cardiomyopathy.20
In the m.3243A>G variant of mitochondrial disease, however, progressive AV-conduction problems rarely occur and so the brady-induced or brady-induced tachycardia arrhythmia mechanisms seen in KSS are unlikely to apply. Of possible relevance to the m.3243A>G phenotype are the rare reports of polymorphic ventricular tachycardia occurring in patients with KSS in the absence of abnormal AV-conduction, bradycardia or QTc-interval prolongation.8 This scenario is more akin to the context of patients with the m.3243A>G phenotype. The mechanism can be explained in either mitochondrial variant by the development of sufficient myocardial fibrosis to make scar re-entrant arrhythmias possible. In mitochondrial patients with normal AV-conduction, extensive myocardial fibrosis on cardiac MRI might prove useful in identifying those at higher risk of lethal arrhythmias.7 12
The implication of not finding a recurring pattern of ‘warning arrhythmias’ is this study may also point to rarer arrhythmia mechanisms, allowing lethal arrhythmias to only occur when transient other factors interact with the, typically mild, degree of ventricular hypertrophy and minimal fibrosis.8 10–12 Such scenarios have been thought particularly likely in patients with extensive respiratory chain deficiency in the heart.21–25 Arrhythmias might be triggered, for example, during episodes of sepsis,26 lactic or keto-acidosis, metabolic or electrolyte derangement23 27 or with medication, toxin or alcohol ingestion.24 28–30
There is also the alternative possibility that some SD in the m.3243A>G mitochondrial disorder are not primarily due to cardiac arrhythmias at all.30 SD in some patients with tonic-clonic forms of epilepsy31 32 have been shown to occur due to prolonged, post-ictal hypoxia and brain-stem dysfunction with asystole occurring terminally.33 34 A similar scenario might also explain some SDs in patients with mitochondrial disease, during epileptic or encephalopathic crises. Implanted ECG-loop recorders could be deployed in further studies to explore whether rhythm-events occur selectively at these times.
Limitations
The study has a number of obvious limitations. The number of participants studied was small and may not represent the wider group from which they were drawn. Although recruited sequentially on the basis of having a pre-defined, perceived ‘high risk’ mtDNA variant for SD, participants were at varying stages in the evolution of their mitochondrial phenotype and none had abnormal AV-conduction, WPW syndrome or experienced stroke-like episodes during follow-up. Six of the nine patients studied were taking beta-blocker therapy which might have prevented arrhythmias. Only two subjects were assessed for the presence and extent of myocardial fibrosis. The burden of non-sustained, ventricular arrhythmias might differ significantly between those with and without intra-myocardial scaring, despite having similar measures of ventricular function and hypertrophy on echocardiography. Perhaps most importantly, the negligible prevalence of arrhythmias observed cannot exclude categorically the possibility that these patients were capable of arrhythmias that could cause SD under specific conditions.
Conclusions
Patients in one of the perceived higher ‘risk groups’ for SD from the m.3243A>G mitochondrial disorder had negligible prevalence of arrhythmias during prolonged ECG surveillance. This suggests that the myocardium in the m.3243A>G disorder is not primarily arrhythmogenic. It was always unlikely that, the typically mild, degree of ventricular hypertrophy or dysfunction in most mitochondrial patients with this phenotype could provide sufficient substrates for tachy-arrhythmias. The main implication of our results is that, the mechanism of SD in these patients remains unknown and may not be primarily arrhythmic. If arrhythmias are responsible, they can probably only occur when a range of dynamic, proarrhythmic factors interact transiently.
Data availability statement
All data relevant to the study are included in the article or uploaded as online supplemental information.
Ethics statements
Patient consent for publication
Ethics approval
This project was justified clinically on the basis of our earlier conclusions about the risk and prevalence of sudden death in patients with particular mitochondrial genotypes. As such it did not require specific ethics approval.
References
Footnotes
Contributors JB: principal investigator, study concept, design, patient recruitment and data acquisition, study supervision and interpretation of data, drafting manuscript for content, final approval of manuscript, responsible for the overall content as the guarantor for all aspects of the work. YSN: study design and patient recruitment, data acquisition, manuscript preparation, redrafting for content and final approval. MT: device implants and programming, data acquisition and surveillance, manuscript redrafting and final approval. MGDB: study design, data acquisition and recruitment, drafting manuscript for content and final approval. SM: data collection, manuscript drafting for content and final approval. DT: study concept, design, securing funding, manuscript critique and redrafting for content and final approval. GSG: study concept, design, securing funding, patient recruitment, manuscript preparation, review for content and final approval.
Funding This work was supported by the Wellcome Centre for Mitochondrial Research (203105/Z/16/Z), Newcastle University Centre for Ageing and Vitality (supported by the Biotechnology and Biological Sciences Research Council and Medical Research Council L016354), UK National Institute for Health Research (NIHR) Biomedical Research Centre (BRC) for Ageing and Age-Related Disease award to the Newcastle Upon Tyne Hospitals NHS Foundation Trust, NIHR, and the UK NHS Specialist Commissioners, which funds the Rare Mitochondrial Disorders of Adults and Children Diagnostic Service in Newcastle Upon Tyne (http://www.newcastle-mitochondria.com/). This work also received infrastructure support from the UK Mitochondrial Disease Patient Cohort: A Natural History Study and Patient Registry (REC ref 13/NE/0326), NIHR Biomedical Research Centre, Newcastle and North Tyneside Comprehensive Local Research Network. YSN. held an NIHR Clinical Lectureship in Neurology (CL-2016-01-003). We acknowledge gratefully also each of the patients who participated in this project.
Disclaimer The views expressed are those of the authors and not necessarily those of the NHS, the NIHR, the BRC or the Department of Health.
Competing interests In addition to the institutional support from the organisations listed under acknowledgments above, all authors have completed the ICMJE uniform disclosure form with declarations as follows: JB: research grant from Duchenne UK; Trial Data Monitoring Committee member, Sarepta Therapeutics, Cambridge, MA, USA; Consulting fees from EspeRare Foundation, Geneva, Switzerland. YSN: declares no conflicts of interest. MT: declares no conflicts of interest. MGDB: declares no conflicts of interest. SM: declares no conflicts of interest. DT: declares receipt of grants from Wellcome Trust and MRC; consulting fees from IMEL Biotherapeutics, Tokyo, Japan, Nanna Therapeutics, Cambridge, UK and Casma Therapeutics, Cambridge, MA, USA; Trustee and Board Member UK Dementia Research Institute (unpaid). GSG: research awards NIHR BRC, Newcastle upon Tyne and Wellcome Trust.
Provenance and peer review Not commissioned; externally peer reviewed.