Article Text

Abstract
Objective This study summarises the diagnostic validity and clinical utility of genetic testing for patients with hypertrophic cardiomyopathy (HCM) and their at-risk relatives.
Methods A systematic search was performed in PubMed (MEDLINE), Embase, CINAHL and Cochrane Central Library databases from inception through 2 March 2020. Subgroup and sensitivity analyses were prespecified for individual sarcomere genes, presence/absence of pathogenic variants, paediatric and adult cohorts, family history, inclusion of probands, and variant classification method. Study quality was assessed using the Newcastle-Ottawa tool.
Results A total of 132 articles met inclusion criteria. The detection rate based on pathogenic and likely pathogenic variants was significantly higher in paediatric cohorts compared with adults (56% vs 42%; p=0.01) and in adults with a family history compared with sporadic cases (59% vs 33%; p=0.005). When studies applied current, improved, variant interpretation standards, the adult detection rate significantly decreased from 42% to 33% (p=0.0001) because less variants met criteria to be considered pathogenic. The mean difference in age-of-onset in adults was significantly earlier for genotype-positive versus genotype-negative cohorts (8.3 years; p<0.0001), MYH7 versus MYBPC3 cohorts (8.2 years; p<0.0001) and individuals with multiple versus single variants (7.0 years; p<0.0002). Overall, disease penetrance in adult cohorts was 62%, but differed significantly depending on if probands were included or excluded (73% vs 55%; p=0.003).
Conclusions This systematic review and meta-analysis is the first, to our knowledge, to collectively quantify historical understandings of detection rate, genotype-phenotype associations and disease penetrance for HCM, while providing the answers to important routine clinical questions and highlighting key areas for future study.
- cardiomyopathy, hypertrophic
- systematic reviews as topic
- genetic diseases, inborn
Data availability statement
Data are available upon reasonable request. Data are available from Susan Christian at smc12@ualberta.ca.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Key questions
What is already known about this subject?
As one of the most common inherited conditions, hypertrophic cardiomyopathy (HCM) is a routine indication for genetic testing. However, our understanding of the impact of genetic testing on clinical outcomes has been limited to individual studies or small analyses until now,
What does this study add?
In this systematic review and meta-analysis, historical understandings of HCM from across 25 years are collectively quantified. Detection rate based on pathogenic and likely pathogenic variants was highest in paediatric cohorts and adults with a positive family history. Application of current, improved, variant interpretation standards significantly impacted the adult detection rate of gene panel testing. Age-of-onset in adults was significantly earlier for genotype-positive cohorts and those with MYH7 or multiple variants. Overall, disease penetrance was 62%, but differed significantly depending on if probands were included or excluded.
How might this impact on clinical practice?
A refined understanding of genetic testing validity and clinical utility for HCM provides critical clinical information to guide and optimise management for patients and at-risk relatives.
Introduction
Hypertrophic cardiomyopathy (HCM) is characterised by left ventricular hypertrophy in the absence of predisposing cardiac conditions, most commonly inherited as autosomal dominant, and has a prevalence of 1/500.1 Since the first pathogenic variant for HCM was discovered in 1990,2 numerous studies have individually addressed genetic testing for HCM and current professional guidelines recommend genetic testing for affected individuals and their at-risk relatives.3 4 While these recommendations primarily focus on the benefits of cascade genetic testing for at-risk relatives, permitting early diagnosis and risk stratification for sudden cardiac death (SCD), the direct benefits for patients with HCM are less clear. The objective of this systematic review was to assess the diagnostic validity and clinical utility of genetic testing for patients with HCM and at-risk relatives.
Methods
A systematic review was performed to align with the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA)5 reporting checklist to address the overarching research question, ‘Does genetic testing lead to improved outcomes for individuals diagnosed with HCM and their at-risk relatives?’ This question has several components, including the detection rate for gene panel testing, genotype–phenotype correlations, penetrance and management implications, which are reported in this manuscript. Additional questions relating to uptake, utility and patient-reported outcomes for genetic testing and genetic counselling are detailed in a second manuscript that has been submitted for publication.
The research team, consisting of medical librarians, a methodologist and genetic counsellors, defined the PICOTS (population, interventions, comparators, outcomes, timing and setting), which are presented in online supplemental methods table 1. A search strategy was developed using keywords pertaining to HCM, genetic counselling and genetic testing. We queried the PubMed (MEDLINE), Embase, CINAHL and Cochrane Central Library databases with minor modifications to accommodate the search input parameters for each database. The initial search was conducted on 7 July 2017 and updated on 2 March 2020. The PubMed (MEDLINE) search strategy is presented in online supplemental methods table 2. Articles were limited to English-language publications.
Supplemental material
Summary of detection rate analyses
Summary of genotype–phenotype analyses in predominantly adult cohorts
All phases of the review and extraction process were performed in duplicate by blinded reviewers, and disagreements were adjudicated through discussion, or with the aid of a third reviewer. Deduplicated citations were uploaded to Rayyan6 for abstract and full-text review according to prespecified inclusion and exclusion criteria based on the PICOTS (online supplemental method table 3). Outcome-specific exclusion criteria are reported in online supplemental method table 4. Studies identified in the updated literature search were screened and reviewed in their entirety in Covidence. Relevant data were extracted into an Excel spreadsheet by reviewers. Study quality was assessed using the Newcastle-Ottawa tool.7
Summary of penetrance analyses
Data analysis
We prespecified the analysis plan and data were grouped into three main categories: detection rate, genotype-phenotype correlations and penetrance. Data analysis, including generation of forest plots, was performed using R V.4.0.2 with ‘meta’, ‘metafor’ and ‘stats’ packages. Meta-analysis of single proportions was calculated with generalised linear mixed model, random-effects settings.8 Continuous variables and multiple proportions were assessed using inverse variance, random-effects meta-analyses. Because genes tested included those with definitive, strong, moderate and weak associations to HCM, further subgroup analysis was limited to the eight sarcomeric genes with definitive association to disease (ACTC1, MYBPC3, MYH7, MYL2, MYL3, TNNT2, TNNI3 and TPM1).9 In addition, subgroup and sensitivity analyses were performed for genotype-positive (G+) versus genotype-negative (G−) patients, inclusion of probands in study population, paediatric and adult cohorts, family history and variant classification standard used. In studies not reporting the unique number of patients with a family history of either SCD or cardiomyopathy (CM), we included the largest reported group (either SCD or CM history) in our meta-analysis of detection rate, to avoid double-counting patients and inflating the pooled estimate. Studies not included in the meta-analyses were narratively synthesised and their results were compared with the meta-analysis results.
Between-group comparisons were calculated with the appropriate statistic (eg, χ2) for articles that presented their data alternatively, where possible.10–13 Meta-analyses are reported as the pooled estimate with accompanying CIs and p values for between-group comparisons. Heterogeneity was calculated as I2 and τ2 and is reported on the accompanying forest plots. Significance was set at p<0.05; no adjustment was made for multiple comparisons.
Results
A total of 3196 non-duplicated articles were screened and 596 were reviewed in their entirety for inclusion. Data extraction and quality assessments were performed on 132 articles meeting inclusion criteria (online supplemental figure 1). In total, 80 studies reported on detection rate, 44 described genotype–phenotype associations and 51 provided penetrance estimates (categories not mutually exclusive). No studies reporting on management implications were identified. Online supplemental table 1 provides a summary of all studies and more comprehensive data are provided in online supplemental tables 2-12.
Detection rate
Detection rate (table 1) was evaluated in predominantly adult and paediatric cohorts (online supplemental tables 2-5). The detection rate was based on both pathogenic and likely pathogenic variants, as defined per publication. Subgroup data analyses were based on the application of American College of Medical Genetics and Genomics (ACMG) and Association for Molecular Pathology (AMP) variant classification standards, relevant family history, presence of multiple variants and gene prevalence.9 14 In addition, utilisation of exome and genome sequencing in HCM cohorts is described.
Adults
The pooled detection rate in predominantly adult HCM cohorts was 42% (figure 1) with an inconclusive rate (ie, rate of results with ≥1 variants of uncertain significance) of 12% (online supplemental figure 2). Studies that applied current ACMG/AMP standards had a lower detection rate than those that did not (33% vs 43%; p=0.0001), and a higher inconclusive rate (24% vs 10%; p<0.0001). Identification of two or more disease-causing variants was reported in 2% of cases (online supplemental figure 3).
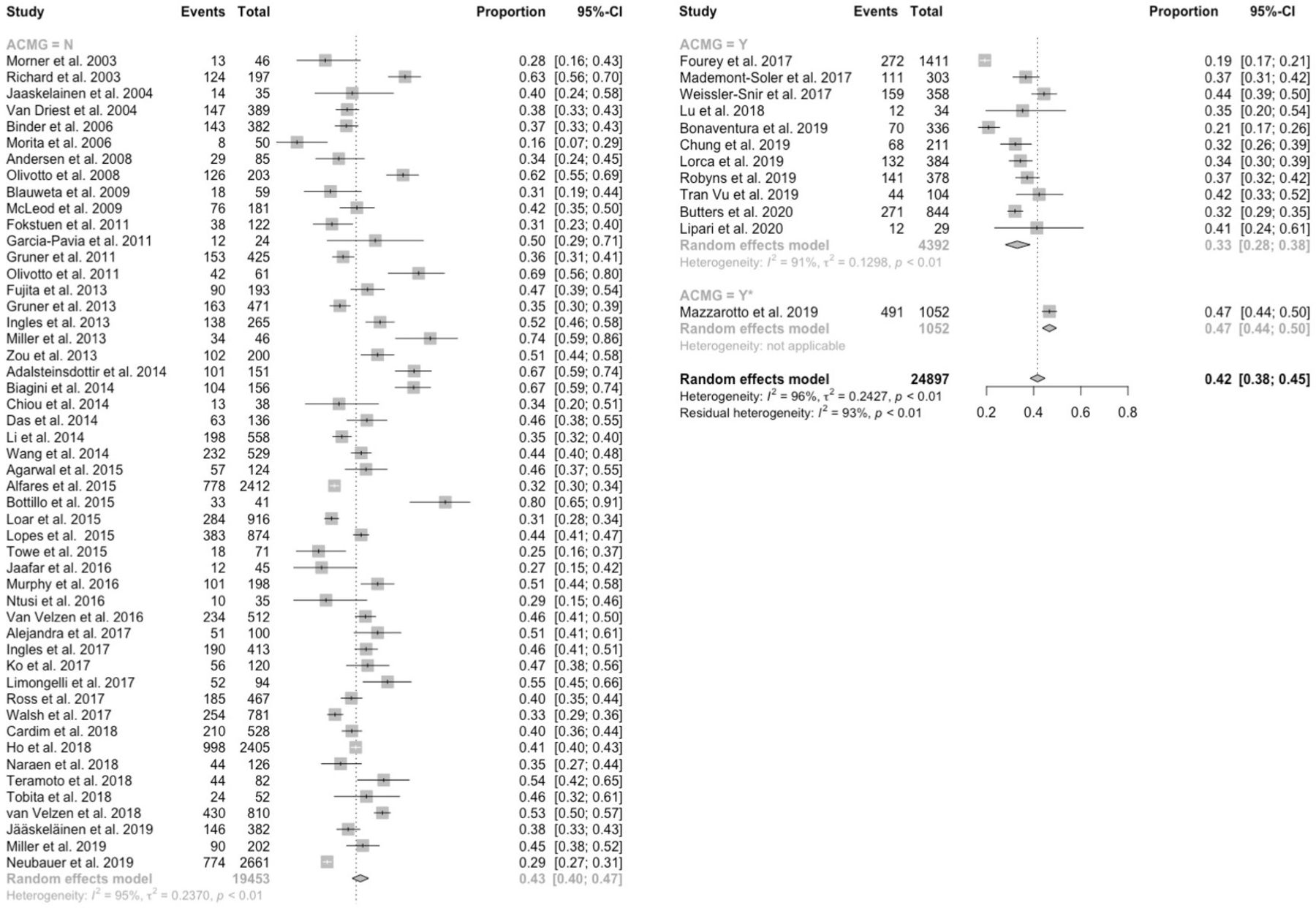
Forest plot of detection rate in predominantly adult HCM cohorts by usage of the ACMG/AMP standards. The pooled detection rate was 42%. Studies that applied ACMG/AMP standards had a lower detection rate than those that did not use ACMG/AMP standards (33% vs 43%; p=0.0001). ACMG/AMP, American College of Medical Genetics and Genomics/Association for Molecular Pathology; HCM, hypertrophic cardiomyopathy.
The detection rates for adult HCM cohorts with a positive family history of HCM (61%), SCD (57%) or CM±SCD (59%) were significantly higher compared with apparently sporadic cases (33%; between-group comparison p=0.005; online supplemental figure 4).
The majority of individuals with a positive result (96%) had at least one disease-causing variant identified in one of the eight sarcomeric HCM genes, and MYBPC3 and MYH7 were collectively the most commonly observed among positive results (81%; online supplemental figure 5).
Pediatrics
The pooled detection rate in paediatric HCM cohorts (≤21 years old) was 56% (online supplemental figure 6) with an inconclusive rate of 19%–31%. The detection rate for paediatric cohorts was significantly higher compared with the predominantly adult cohorts (56% vs 42%; p=0.01; online supplemental figure 6). Studies that applied current ACMG/AMP standards had a higher detection rate than those who did not (78% vs 52%; p<0.0001). Identification of two or more disease-causing variants was reported by two studies as 5% and 6% of cases, which was not significantly higher compared with adults (5% vs 2%; p=0.06; online supplemental figure 3).
The detection rate for paediatric HCM cohorts with a positive family history (HCM: 58%; SCD: 48%; or CM±SCD: 57%) did not differ significantly from either sporadic cases (49%) or the overall detection rate unselected for family history (56%; between-group comparison p=0.49; online supplemental figure 7).
Similar to adult cohorts, variants in the eight sarcomeric HCM genes (62%–97%), as well as MYBPC3 and MYH7 (59%–96%), accounted for the majority of positive results.
Exome/genome sequencing
Few studies that met our inclusion criteria reported on the detection rate of exome (n=3) and genome (n=2) sequencing. Results are summarised in online supplemental table 4. Seidelmann et al15 and Mak et al16 identified disease-causing variants from exome sequencing in 46% and 43%, respectively. Comparatively, Nguyen et al17 performed exome sequencing on 200 individuals with HCM and found variants in 88%, though the majority were in genes other than the eight sarcomeric HCM genes and limited information was provided on the variant classification approach. Two studies of genome sequencing directly compared findings against other testing methods.18 19 Cirino et al18 identified 19 of 20 variants previously found by panel testing and 1 pathogenic variant in a previously negative case. Bagnall et al19 identified disease-causing variants in 9 of 46 cases (20%) with previously negative genetic testing, including four in genes not previously tested and four deep intronic splice variants in MYBPC3.
Genotype–phenotype implications for prognosis
Analyses focused on genotype–phenotype associations for age-of-onset, sudden cardiac arrest (SCA), presence of an implantable cardioverter–defibrillator (ICD), heart failure (HF), septal reduction therapy and mortality. Genotype comparisons included: genotype-positive (G+) versus genotype-negative (G−), MYBPC3 versus MYH7 and multiple versus single variants (table 2; online supplemental tables 6-11).
Age-of-onset
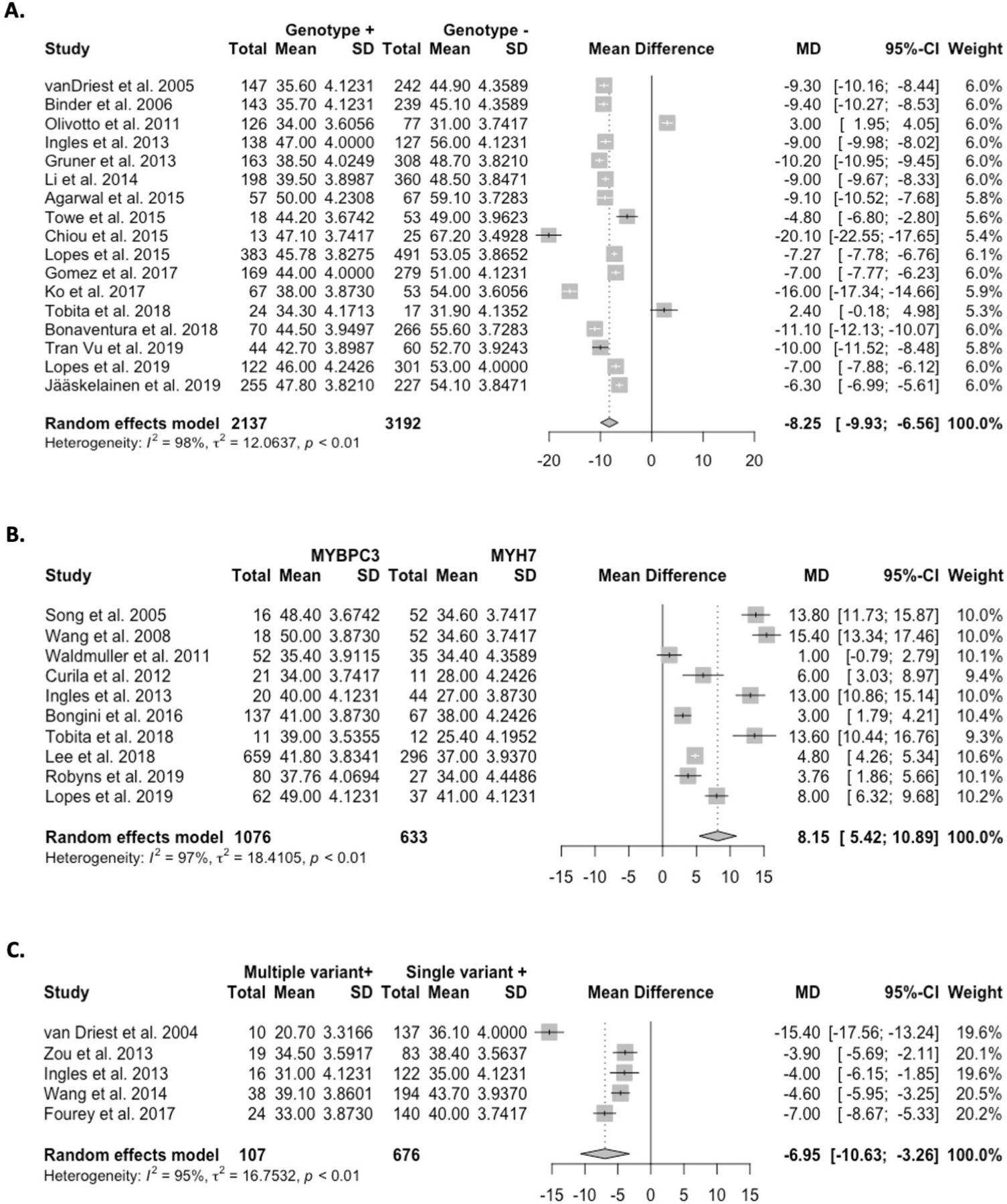
The pooled mean age-of-onset in predominantly adult cohorts was 8.3 years earlier for G+ versus G− cohorts (p<0.0001; figure 2A). One additional study reported median age-of-onset and similarly found that G+ individuals were younger at disease onset (50 years vs 59 years).20 Comparatively, three paediatric studies did not observe differences.12 21 22
{kind=link}
{kind=link}
Forest plots of age-of-onset in predominantly adult cohorts across (A) genotype-positive (G+) vs genotype-negative (G−) cohorts; (B) MYBPC3 vs MYH7 cohorts; and (C) multiple vs single variant cohorts. The pooled mean age-of-onset was 8.3 years earlier for G+versus G− individuals (p<0.0001), 8.2 years later for variants in MYBPC3 vs MYH7 (p<0.0001), and was 7.0 years earlier for individuals with multiple variants versus those with a single variant (p<0.0002).
The pooled mean age-of-onset in adult cohorts was 8.2 years later for variants in MYBPC3 versus MYH7 (p<0.0001; figure 2B). Two additional studies reported median age-of-onset and findings were consistent with the meta-analysis.23 24 Two paediatric studies found no significant difference in age-of-onset for MYBPC3 cohorts compared with MYH7 cohorts.21 25
The pooled mean age-of-onset in adults with multiple variants was 7.0 years earlier than those with a single variant (p<0.0002; figure 2C). One study reporting median ages-of-onset also found that multiple variants were significantly associated with an earlier age-of-onset.23 Findings from two paediatric studies were discordant.21 25
Sudden cardiac arrest
SCA was defined as resuscitated cardiac arrest, SCD, appropriate ICD therapy or a combination of these events. Kaplan-Meyer analysis in a British study (n=874) and a Portuguese registry (n=422) found that G+ individuals were significantly more likely to experience SCD compared with G− individuals (p=0.03 and p=0.02, respectively).26 27 Although a similar trend was seen in our meta-analysis of five studies comparing G+ versus G− cohorts, the OR of SCA (OR 1.4; online supplemental figure 8) did not reach statistical significance. Finally, the hazard ratio (HR) determined by van Velzen et al28 (HR 1.0; 95% CI 0.6 to 1.9) did not suggest a difference in SCA between groups.
Meta-analysis of six studies that compared SCA in MYBPC3 versus MYH7 cohorts (online supplemental figure 8) found no significant difference between groups (OR 0.9), consistent with HRs from a large registry study.29
Findings from four adult studies comparing SCA in cohorts with multiple versus single variants were mixed; two studies found no significant difference, whereas two studies reported a higher incidence of SCD in individuals with multiple variants.11 29–31 The one paediatric study did not report a significant difference.32
ICD implantation
ICD implantation was more common in G+ cohorts than G− cohorts in an analysis of 10 studies (OR 1.9; p<0.0001; online supplemental figure 9). However, the same comparison in two paediatric studies did not find a significant difference between groups.12 21 No significant difference was found across six studies of adults comparing MYPBC3 versus MYH7 cohorts (OR 1.2; online supplemental figure 9) nor in two studies of adult cohorts with multiple versus single variants.13 30 However, one paediatric study reported a significantly higher hazard of ICD implantation in individuals with multiple variants (HR 4.4; 95% CI 1.8 to 11.0; p<0.001).32
Heart failure
HF outcomes included New York Heart Association (NYHA) class III/IV, cardiac transplantation, left ventricular ejection fraction, HF admissions and HF symptoms. No significant differences were observed in NYHA class outcomes when comparing G+ versus G−, MYBPC3 versus MYH7 or multiple versus single variants (online supplemental figure 10). One large registry-based study that found individuals with MYH7 variants more likely to require cardiac transplant or ventricular assist device (VAD) than individuals with MYBPC3 variants (HR 2.8; 95% CI 1.3 to 5.8) and individuals with multiple variants were more likely to require cardiac transplant (HR 7.5; 95% CI 2.7 to 20.5).29 32
Septal reduction therapy
Septal reduction therapy included myectomy, ablation or a combined outcome of myectomy and/or ablation. No significant differences were observed in these outcomes when comparing G+ versus G−, MYBPC3 versus MYH7 or multiple versus single variants (online supplemental figure 11).
Mortality
Mortality was reported as death, all-cause mortality, cardiac mortality, HCM-related death and survival. While there were discordant findings, the majority of studies found no significant difference in mortality across seven studies comparing G+ versus G− cohorts23 26 28 33–36 and seven studies comparing MYBPC3 versus MYH7 cohorts.10 24 29 36–39 Findings were split when comparing individuals with multiple versus single variants with two adult studies showing a significant difference, while one adult and one paediatric study did not.23 29 30 32
Disease penetrance
The penetrance of HCM disease-causing variants was evaluated in 51 predominantly adult and paediatric cohorts that both included and excluded probands, and at a gene-specific level (table 3; online supplemental table 12). The pooled penetrance of HCM across adult cohorts was 62%. Overall penetrance differed significantly depending on if probands were included or excluded (73% vs 55%; p=0.003; online supplemental figure 12).
Pooled penetrance in paediatric cohorts (<20 years old) was 24% across five studies (online supplemental figure 13). The penetrance in a paediatric cohort with multiple variants that included probands was 81%.32
Gene-specific penetrance in adult and paediatric cohorts was 65% for MYBPC3, 76% for MYH7 and 77% for TNNT2 (online supplemental figure 14). Two studies reported penetrance for TNNI3 (48% and 56%) and ACTC1 (89% and 100%).40–43 Additional genes were limited to a single study.
Three studies reported a higher disease penetrance in men.44–46 Two of which presented age-based penetrance for each sex: one included probands and penetrance by age 40 years was 92% for men and 67% for women45; the other excluded probands and penetrance by age 40 years was 77% for men and 35% for women.46 The mean age at study enrolment and of disease onset was only reported in 39% and 22% of cohorts, respectively, limiting the opportunity to further evaluate age-based penetrance. The oldest mean cohort age was 57 years with the majority of cohorts having a mean age in the 40s.
Discussion
This systematic review and meta-analysis summarises and quantifies data on HCM detection rate, genotype–phenotype associations and disease penetrance from 132 publications across 25 years, confirming several well-reported trends and previously established associations.
Detection rate
Numerous studies have published on the detection rate of HCM genetic testing and meta-analysis of these data demonstrate that the yield of pathogenic and likely pathogenic variants is influenced by multiple factors. Consistent with traditional convention and prior systematic reviews and meta-analyses of adult cases with HCM, a relevant family history significantly increases detection rate.47 48 Alternatively, while paediatric cases have a significantly higher detection rate compared with adults, family history does not significantly alter the detection rate.
Approaches to variant classification are an established variable to these analyses, but improvements in variant classification have also impacted detection rate with the 2015 ACMG/AMP standards being considered the most accurate guidelines in North America.14 Not surprisingly, adult studies that applied the ACMG/AMP standards had a lower overall detection rate based on pathogenic and likely pathogenic variants and higher inconclusive rate (variants of uncertain significance), likely representing a more accurate estimate of current detection rates. Interestingly, when looking at the same comparison for paediatric cases, the two studies that applied ACMG/AMP standards had a significantly higher detection rate, despite not finding any notable differences in these cohorts. Given that these findings are counter-intuitive and the number of paediatric studies that applied ACMG/AMP standards was limited, additional research is needed.
Expanding to exome/genome sequencing
With exome and genome sequencing for HCM increasingly available, studies have found that most disease-causing variants remain identifiable by large gene panels.15–17 Technical differences between exome/genome sequencing and traditional gene panels remain an important consideration, potentially impacting the sensitivity for some genes.16 A recent example is deep intronic and other non-coding variants identifiable by genome sequencing, but missed by traditional panels and exome sequencing, though additional evidence supporting pathogenicity is needed (eg, segregation analysis, functional studies, additional case data).19 49
Genotype–phenotype implications on outcomes
Analyses of genotype–phenotype associations focused on three comparisons: genotype-positive (G+) versus genotype-negative (G−), MYBPC3 versus MYH7 and multiple versus single variants. A significant difference in age-of-onset was observed in all three comparison groups, as has been reported previously,47 supporting that genotype influences age-of-onset in HCM. However, while Lopes et al did not observe a significant difference between MYBPC3 and MYH7, our analysis included six additional studies. In a more recent review, Sedaghat-Hamedani et al approached their meta-analysis differently by looking at individuals across studies who were G−, MYBPC3 + and MYH7 + and concluded that age of onset was earliest for MYH7 + individuals.50
Although a significantly higher rate of SCA was reported for G+ individuals by two large studies, the association did not reach significance in our meta-analysis. The large multisite Sarcomeric Human cArdiomyopathy REgistry (SHaRe) study (n=2763) reported a similar association but was excluded from our analysis of G+ versus G− individuals as not all genotype-negative individuals had genetic testing for all eight sarcomere genes.29 Furthermore, in the meta-analysis by Sedaghat-Hamedani et al, they conclude that G+ individuals have a higher rate of SCA compared with G− individuals.50
We found that G+ individuals were more likely to have an ICD. While this raises the possibility that genotype status influences ICD utilisation, family history may contribute since individuals with a family history of SCA (a factor considered in SCA risk stratification) are also more likely to carry a disease-causing variant. Similarly, Ingles et al found that individuals with a negative family history are less likely to have an ICD.51
We found no significant differences between genotype across other outcomes, which contrasts prior findings supporting that genotype status and the gene involved are predictive of worse outcomes.29 50 However, this is likely in part due to limitations of how the existing data were categorised and the ability to directly compare across studies. Alternatively, these findings may suggest that genotype is only one of several predictors influencing phenotypic outcomes. More standardised research comparing outcomes across multiple potential predictors is required.
Disease penetrance
Determination of disease penetrance is challenging due to the possibility of selection bias in the included studies. While use of unselected populations would be the most informative, current studies are limited to those that included probands (73%; likely provide an overestimate) versus those that exclude probands (55%; likely provide an underestimate). Because of this, both approaches were considered and the overall disease penetrance across studies was 62%. It should be noted, however, that the average age of most cohorts was in the 40s, limiting the follow-up time and as such the conclusions that can be drawn. Our findings support that MYBPC3 and TNNI3 variants have lower penetrance compared with MYH7 and TNNT2, and penetrance is higher in men, irrespective of the gene, as has been previously reported.47 50
Future research is needed to assess the impact of additional environmental and genetic modifiers on disease penetrance. Although there is evidence to suggest that male sex, obesity, hypertension and exercise could increase penetrance, additional studies are needed.52 Furthermore, polygenic risk scores and a greater understanding of epigenetic factors may further elucidate the risk of disease within and across families.
While beyond the scope of this review, data on penetrance in unselected populations will accumulate over time as the HCM genes are recommended to be reported as medically actionable secondary findings from diagnostic exome and genome sequencing by the ACMG.53 As one example, van Rooij et al found that only 22% of unselected individuals with HCM disease-causing variants showed convincing evidence of disease over 25 years of follow-up.54 Future assessments of HCM disease penetrance should consider presentation of disease in probands, at-risk relatives and unselected patients.
Finally, penetrance during childhood ranged from 7% to 61% with many cases presenting prior to 12 years old. Outcomes from these cohorts may be skewed as a result of the most severe paediatrics cases coming to medical attention. While older guidelines recommend that cardiac screening begin around 10–12 years of age for children with a first-degree relative with HCM, newer guidelines recommend cardiac screening for children under 5 years.55 56 Our findings are consistent with newer recommendations aiming to identify all individuals with disease onset in childhood.
Points of consideration and future research
This study identified multiple limitations that impacted the ability to analyse data across studies, including variability in study design and reporting of outcomes. The predominant study design was observational case series and some outcomes of interest were not the primary focus. With regards to detection rate, variability included the genes tested, methodology used and the variant classification standards applied. The impact of the ACMG/AMP standards was assessed for detection rate; however, it was not evaluated for the other outcomes assessed and, therefore, the impact on these areas remains unclear.
For the analysis of genotype–phenotype associations, there was variability in study design, and in how outcomes were defined and reported. Outcomes related to cardiac events such as SCA were limited to meta-analysis of ORs rather than rates due to how the data were reported by the majority of studies (eg, the studies did not report on time-to-event risks/rates). Our meta-analysis also focused on studies that performed head-to-head comparisons between genotypes, which differs from the meta-analysis performed by Sedaghat-Hamedani et al that evaluated genotypes across studies.50 When considering penetrance, limitations included unreported or younger mean age of the cohorts, limited follow-up time and variability in the proportion of at-risk relatives included in analysis. Often the evaluation of relatives was not the primary focus of the study and, therefore, very limited demographic data were provided.
Finally, there is a specific need for collaborative efforts to standardise approaches. In particular, the areas identified by this systematic review include consistent definitions and reporting on cohorts (both probands and at-risk relatives) and cardiac outcomes, reporting the minimum genetic analyses completed and describing the classification of disease-causing variants. Increased consistency in how results are reported and interpreted would improve transparency and allow for more direct comparisons across studies.
Conclusions
As one of the most common inherited conditions, HCM has long been the focus of research and clinical interest. This systematic review and meta-analysis is the largest for any particular outcome and the first, to our knowledge, to collectively refine and quantify historical understandings of detection rate, genotype–phenotype associations and disease penetrance for HCM. Although the variabilities in study design and reporting of outcomes limited the analyses that could be performed, the large amount of data evaluated provide answers to important routine clinical questions, particularly those related to detection rate and genotype/phenotype correlations. Key areas for future research include expanding genotype–phenotype associations and disease penetrance estimates across various populations. While additional studies are needed, our current analyses serve as an important stepping stone to understanding the clinical utility of genetic testing for a condition that impacts so many families.
Data availability statement
Data are available upon reasonable request. Data are available from Susan Christian at smc12@ualberta.ca.
Ethics statements
Patient consent for publication
Acknowledgments
Support for Jennifer Malinowski’s time was provided by the National Society of Genetic Counselors (NSGC). We want to thank the NSGC Practice Guidelines Committee for identifying the need for this systematic review and supporting this project. The authors would also like to acknowledge the medical librarians at each institution for assisting with the literature searches and to Talin Boghosian for contributing to preparation of the manuscript and the review process.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors All authors contributed to the planning, conduct, drafting and reporting of the work. The manuscript was revised and approved by all authors. SC and MK are responsible for the overall content as guarantors.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests JM is a contract methodologist for the NSGC. AM is a paid consultant for Concert Genetics.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.