Article Text

Statistics from Altmetric.com
Introduction
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is a novel coronavirus first identified in an outbreak of pneumonia in Wuhan, Hubei Province, China in December 2019. Despite mitigation of the initial epidemic, the viral syndrome now named coronavirus disease (COVID)-19 has since spread rapidly throughout the world, representing a pandemic with profound implications for human morbidity and mortality. In turn, the capacity of diverse healthcare and economic systems to cope with rising infections and associated intensive care requirements is strained.
The most extensive clinical experience of this virus to date comes from the more than 80 000 positive cases identified in Hubei Province.1–4 These early clinical reports clearly indicated that although most clinical manifestations of COVID-19 requiring hospitalisation are respiratory, there is a substantial minority of patients who undergo progressive and severe cardiovascular compromise.1 2 4 Subjects at highest risk of death appear to be more elderly patients with pre-existing cardiovascular disease and/or classical risk factors that accompany advanced cardiovascular illness.3 The goal of this viewpoint article is to examine whether the nature of SARS-CoV2 infection and the homeostatic status of high-risk cardiovascular patients can be linked in a mechanistic framework that provides insights into corrective therapy over and above current and emergent antiviral approaches to COVID-19.
There are a number of reasons why COVID-19 may be associated with cardiovascular complications that include inter alia-specific aspects of SARS-CoV-2 structure and receptor targeting, and target cell location and its relationship with cardiovascular disease homeostasis. In addition, there are potential risk amplifiers including hitherto unanticipated interactions between viral targets within the host and established homeostatic pathways that may already be perturbed in patients with advanced cardiovascular disease.
In this viewpoint, we underscored specific alterations in renin angiotensin system-ACE 2 (RAS-ACE2) homeostasis that may contribute to more adverse COVID-19 outcomes in patients with pre-existing cardiovascular disease. Moreover, we proposed and developed a rationale for specific therapeutic interventions that might mitigate some of the more deleterious cellular pathology and cardiovascular effects of this syndrome.
Structure of SARS-CoV-2, viral infection, cell targets and ACE2
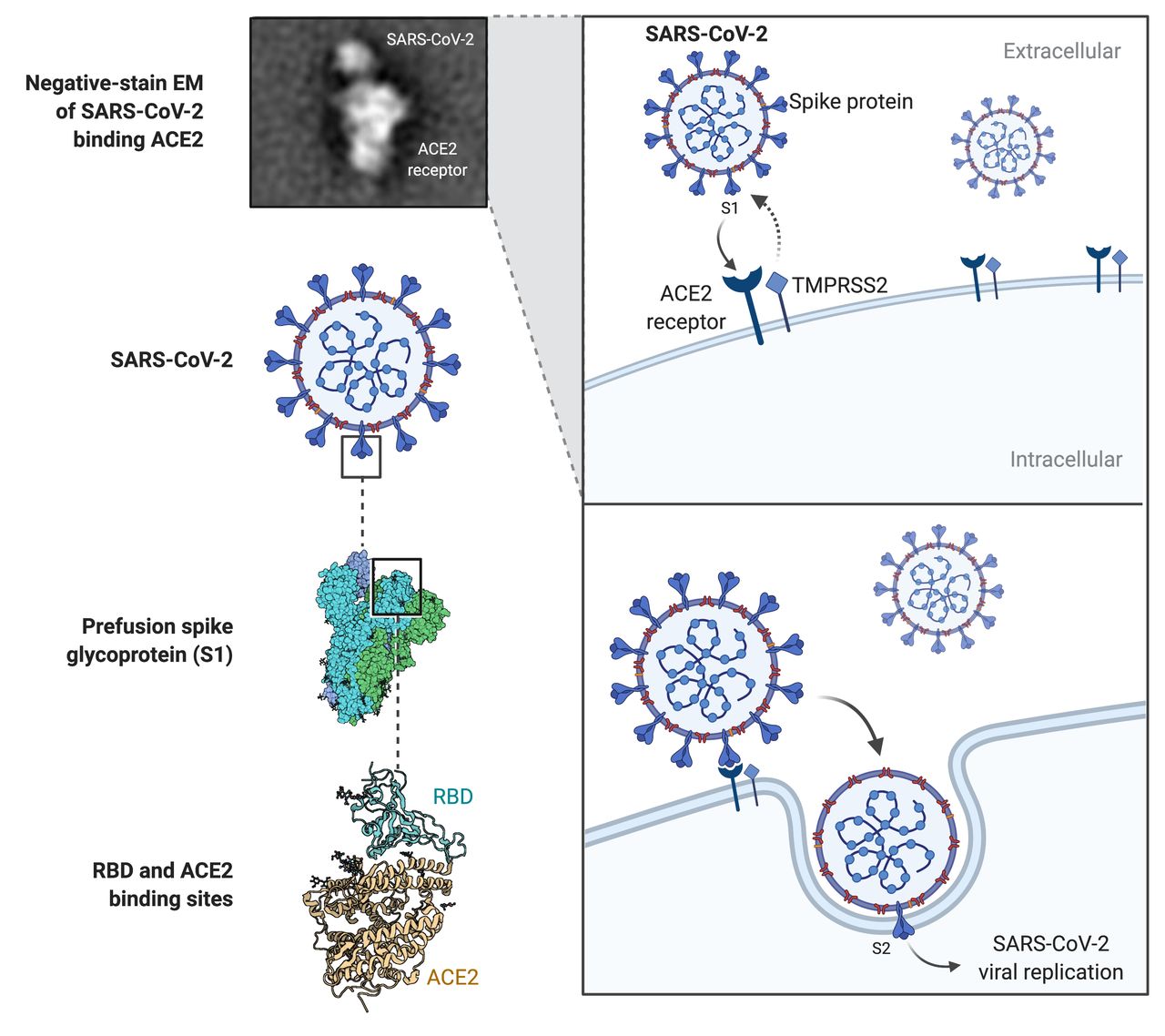
SARS-CoV-2, a positive-strand RNA virus, shares 80% genomic homology with SARS-CoV virus and initial infection is mediated through interaction of virion spike glycoprotein (S protein) with the ACE2 receptor on target cells. The S protein is cleaved into S1 and S2 subunits that act cooperatively to allow ACE2 receptor engagement and viral cell entry.5 S1, via a receptor-binding domain (RBD), binds the peptidase domain of ACE2 with the S2 subunit implicated in membrane fusion.6 S2 cleavage site activation by host transmembrane serine protease 2 (TMPRSS2) is facilitated by a conformational change secondary to S1–ACE2 binding (figure 1).7 Comparative genomic analysis of SARS-CoV-2 suggested that it is optimised for binding to human ACE2 and in this way it exploits the membrane-bound receptor in host cells to initiate and spread infection.7 8

Schematic of SARS-CoV-2 targeting of ACE2 receptor and entry in infected cell. Note cooperative interaction between both viral subunits S1 and S2 and cell surface-bound ACE2 and transmembrane serine protease 2.5–7 EM, electron microscopy; RBD, receptor-binding domain; SARS-CoV-2, severeacute respiratory syndrome coronavirus 2; TMPRSS2, transmembrane serine protease 2.
The primary role of ACE2 in health is maturation of angiotensin, a peptide implicated in vascular homeostasis, vasomotor tone and blood pressure regulation.7 Importantly ACE2 is expressed on diverse human cells including epithelial cells in the lung and small and large intestines, tubular cells of the kidney, vascular endothelial and smooth muscle cells and cardiomyocytes (figure 2)9 and reduction in ACE2 is well known to be associated with hypertension, diabetes, coronary artery disease, myocardial infarct repair and heart failure.10

Multiple human host tissues that express ACE2 receptor highlighting lungs, heart and vasculature. Note clinical consequences of viral infection in cardiovascular and respiratory tissue.1–4 ARDS, acute respiratory distress syndrome.
Although robust histopathological data are still unavailable for organ and tissue damage in COVID-19 patients, it may be useful to look at the homologous SARS-CoV infection that shows similar ACE2 viral tropism. Pathological examination of SARS-CoV subjects has identified viral infection throughout the respiratory tract, spleen and lymph nodes, intestinal epithelium and mucosal lymphoid tissue, liver hepatocytes, tubular epithelium of the kidney, neural cells in the brain and cardiomyocytes and vascular cells within the heart.11 Moreover, postviral cell apoptosis, necrosis, interstitial oedema, macrophage infiltration, lymphocyte depletion and early fibrosis have been documented to varying degrees in all of these tissues in SARS-CoV subjects. Preliminary real-time PCR detection of SARS-CoV-2 in different clinical specimens shows similar widespread seeding of virus in respiratory, circulatory and gastrointestinal systems.12 Although the exact mechanism by which SARS-CoV-2 induces cellular damage remains unknown, clinical and laboratory data from early cohort studies1–4 would support a similar tissue distribution to SARS-CoV with a notable increase in clinical cardiovascular effects.11
In naïve humans subjects, SARS-CoV successfully evades the innate immune response and hijacks host cell metabolism through initial degradation of host mRNA and modulation of ubiquitination.13 Efficient viral replication within the host cell then ensues, leading to cell damage and viral-induced cytolysis. Interestingly, comparative analysis of two successive SARS epidemics in early 2000s showed that increased affinity of the SARS virus for human ACE2 receptor strongly predicted severity of clinical disease suggesting that spike protein conformation is potentially a key determinant of virulence.14 Moreover, the spike protein present on SARS-CoV and SARS-CoV-2 facilitates host cell-to-cell fusion to form syncytia that may have potential pathological consequences for tissues such as the heart.15 Interestingly, in several Wuhan cohorts cardiac injury and arrythmia were prominent in high-risk COVID-19 subjects.1 3 4 Previous SARS-CoV studies also indicated that clinical and pathological impacts of viral infection in various tissues and organs are determined not solely by viral load or tropism with respect to cell surface receptors but also by the counter-regulatory pathways within the host.16 These host mechanisms include inter alia complex elements of homeostatic and inflammatory pathways, anti-apoptotic responses and the compensatory repair power or functional reserve of tissues and organs within the infected subject. The balance between these countervailing forces in SARS-CoV-2 infection may in large part determine the severity of clinical manifestation and outcome of COVID-19 in human subjects.
ACE2 function, receptor distribution and RAS homeostasis
RAS as a signalling pathway acts as a homeostatic regulator of vascular function, and at a tissue level it regulates function within organs such as the kidney, lungs and heart.17 At a regional level, RAS regulates blood flow to organs and controls trophic responses to a wide range of triggers including cell death, injury, inflammation, fibrosis and vascular repair.18 Many of these regulatory elements involve opposing functions to accommodate rapidly coordinated responses to local stimuli.
The dipeptide carboxypeptidase ACE metabolises angiotensin I to form angiotensin II (AngII) and AngII is subsequently metabolised by the carboxypeptidase ACE2 into the vasodilator angiotensin (1–7) (Ang 1–7).19 While ACE and AngII have been the focus of therapeutic targeting for more than four decades,20 ACE2 has only recently come to prominence as a major player in RAS imbalance, especially in the presence of disease. Although ACE2 shares sequence homology with the extracellular domain of ACE, it functions quite differently as a carboxypeptidase19 and is not blocked by ACE inhibitors. Therefore, ACE2 represents an endogenous counter-regulatory pathway with RAS and acts in opposition to the ACE axis (figure 3). Indeed, in the cardiovascular system ACE2 may be more important than ACE in regulating the local effects of AngII and Ang1–7 with consequences for counter-balancing RAS activation.21 22 AngII contributes to systemic hypertension and locally promotes vasoconstriction, endothelial dysfunction, vascular inflammation, endothelial oxidative stress, smooth muscle cell migration, proliferation and contraction and in situ thrombosis.23 24 In the context of acute respiratory distress syndrome (ARDS), AngII is regarded as a pivotal player in vascular permeability, cytokine amplification and inflammatory cell infiltration.21 23 24 In contrast, Ang1–7 opposes local vascular effects of AngII, has antioxidative and anti-inflammatory effects and attenuates vascular disease in murine models.25

{kind=link}
{kind=link}
{kind=link}
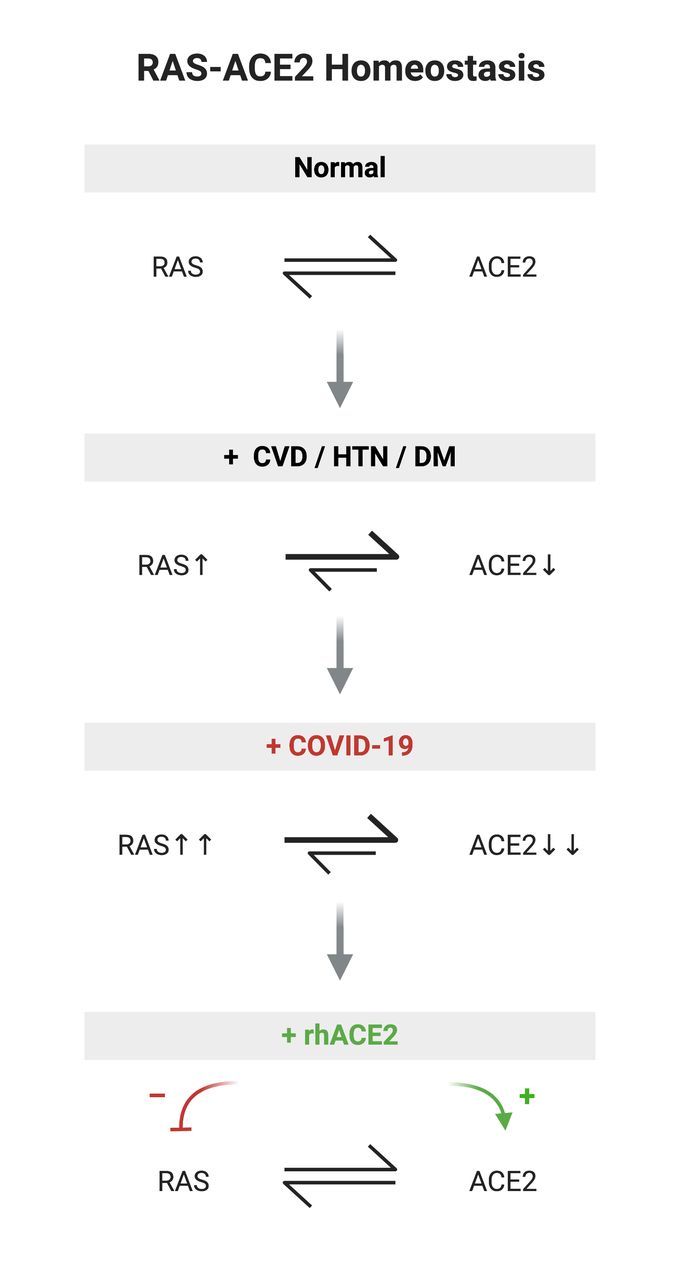
Homeostasis of RAS-ACE2 under normal healthy conditions10 19; perturbation of RAS-ACE2 homeostasis in cardiovascular disease, hypertension and diabetes mellitus22 27; COVID-19 may potentially further upregulate RAS in CVD patients and deplete ACE233; proposition that rhACE2 replacement therapy improves RAS-ACE2 balance by augmenting ACE2 and decreasing RAS activation.37 CVD, cardiovascular disease; HTN, hypertension; DM, diabetes mellitus; RAS, renin angiotensin system; rhACE2, recombinanthuman ACE2.
Genetic knockout studies show that under normal conditions deficiency of ACE2 has relatively modest effects on blood pressure, cardiovascular homeostasis and renal function when compared with ACE gene deletion.26 However, under conditions of oxidative stress, sepsis, inflammation or experimental hypertension, diabetes, atherosclerosis, myocardial infarction or heart failure, ACE2 deficiency greatly amplifies the RAS-associated pathological phenotype in animal models27 and clinical specimens.28 Furthermore, ACE2 knockout models show increased tissue and circulating levels of AngII and reduced Ang1–7,22 with accelerated diabetic nephropathy29 and greater AngII-induced cardiac hypertrophy, fibrosis and infarction,30 whereas ACE deletion confers a modest reduction in circulating AngII, but no effect on tissue levels.31 This would support multiple non-ACE pathways for AngII generation in the body, but limited pathways for AngII degradation, which highlights the potential importance of ACE2 disruption at a tissue level especially in states of RAS activation. Moreover, this underscores the potential of ACE2 replacement as a more potent strategy in reducing AngII and augmenting Ang1–7 compared with conventional RAS inhibition.
Thus, in cardiovascular disease there exists an imbalance between increased RAS activation and reduced ACE2 counter-regulatory effects (figure 3), which may render subjects with increased cardiovascular risk burden susceptible to conditions that worsen asymmetries in RAS/ACE2.
Theoretical implications of ACE2-expressing cell destruction by SARS-CoV-2 in cardiovascular system and other organs
Given that SARS-CoV-2 specifically targets ACE2 receptor and based on SARS-CoV homology and previous SARS-CoV pathology studies, it is reasonable to expect that during viremia host cells and tissues that express ACE2 in the major organs such as lungs, heart, vascular system and kidneys will be susceptible to viral infection.11 32 It is also conceivable that relative loss of ACE2 (through viral destruction) in these affected tissues at a time of increasing RAS activation may act to amplify the pathophysiological consequences of viral infection in these organs. Previous studies of SARS-CoV show viral induction of ACE2 shedding in infected cells33 and downregulation of ACE2 levels in tissues has been linked to viral replication efficiency and pathogenicity.34 Therefore, extensive viral lysis of ACE2-expressing cells may act as a tipping point in tissues ability to adequately respond via RAS/ACE2 to ensuing inflammatory, oxidative stress and ongoing cytokine-mediated cellular insults (figure 3). In contrast, in healthy adults and younger patients, as seen in genetic knockout models,26 it is conceivable that, with RAS-ACE2 homeostasis intact, the consequences of acute viral infection may not reach this threshold.
Two poorly prognostic elements may thus combine in SARS-CoV-2 infection of cardiovascular disease patients, namely (1) a pre-existing imbalance in RAS activity and ACE2 counter-regulation that renders this cohort susceptible to more cardiovascular complications of COVID-19 and (2) destructive targeting by SARS-CoV-2 of ACE2-expressing cells in multiple cardiovascular and other tissues leading to further critical loss of ACE2 reserve. This amplified RAS-ACE2 imbalance in COVID-19 may greatly exacerbate tissue injury and inflammation in affected organs of cardiovascular patients.
Rationale for, and safety of ACE2 replacement therapy in COVID-19 subjects
ACE2 protein replacement has proven beneficial in multiple preclinical models such as hypertension, diabetic vasculopathy and renal disease, postmyocardial infarction, heart failure and especially in ARDS where RAS-ACE2 imbalance is prominent.10 35 36 The mechanism of ACE2 benefit is likely increased metabolism of local tissue AngII and increased bioavailability of Ang1–710 with reduction in reactive oxidation, cell death, inflammation and endothelial cell activation. These protective activities of ACE2 may thus counteract the deleterious effects of tissue injury and associated activation of RAS. In human subjects, this has already been exploited with completion of the first pilot phase 1/2 trial of recombinant human ACE2 (rhACE2) in ARDS.37 In this safety study, which was not powered for efficacy, rhACE2 proved safe and was well-tolerated haemodynamically with a rapid reduction in AngII and increase in Ang1–7 observed after intravenous infusion.37 Pharmacokinetic evaluation of rhACE2 in healthy volunteers supports a twice daily infusion,38 although given a relatively short half-life it is likely that continuous infusion if tolerated would be more effective in more severely ill subjects. AngII and Ang1–7 responses to ACE2 replacement are rapid (within 30 min) so it may be feasible to identify responders and non-responders quickly, although AngII assay variability may require further study and validation.39 Moreover prescreening using AngII levels, ACE2/ACE activity ratios and prognostic ACE gene polymorphisms may further assist in risk stratification of patients more likely to respond to ACE2 therapy.39 We proposed that existing COVID-19 subjects should have an ACE2 deficiency state confirmed prospectively by analysis of ACE2, AngII and Ang1–7 levels in low-risk and high-risk subjects with further dichotomisation based on survival or non-survival. In severe COVID-19, with no current options to save these patients, there is also a rationale for testing the ACE2 deficiency hypothesis in phase 1/2 randomised controlled trials of rhACE2 protein in subjects at high risk of death. Such risk stratification could be based on conventional intensive care unit prognostic scoring systems in addition to circulating factors that indicate poor outcomes such as elevated soluble urokinase plasminogen activator levels.40
We noted a recently planned open-label, randomised controlled trial of rhACE2 for treatment of patients with COVID-19 in Wuhan.41 Further studies will be required to identify whether ACE2 therapy should be targeted only at high-risk cardiovascular patients or all patients at risk of progression to cardiorespiratory failure and death.
Administration of ACE2 recombinant protein in COVID-19 patients raises the question of whether this might exacerbate SARS-CoV-2 infection given viral tropism. All experimental data suggested that the opposite response is more likely. Despite this early phase 1/2 clinical trials should evaluate potential side effects such as hypotension, tachycardia and worsening respiratory failure with close monitoring of vital signs, respiratory parameters, electrocardiography and haematology and blood chemistry profiles. Given that ACE2 downregulation by SARS-CoV is linked to viral replication efficiency,33 34 it is conceivable that ACE2 augmentation may reduce virulence. Importantly, rhACE2 is a soluble protein whereas SARS-CoV-2 binds to cell membrane-bound ACE2 and requires the cooperation of the TMPRSS2 protease to achieve viral cell entry. Therefore, soluble ACE2 alone is likely insufficient to allow viral infection of cells. Previous studies with SARS-CoV and more recently SARS-CoV-2 studies42 showed that soluble ACE2 protein actually inhibits SARS-CoV-2 viral spike RBD binding to membrane ACE2 receptor. For SARS-CoV pathogenesis, ACE2 not only acts as a receptor for viral entry but also protects against local tissue injury.35 If the same holds true for SARS-CoV-2, then soluble rhACE2 may reduce ongoing SARS-CoV-2 access to membrane-bound ACE2 receptor, alter favourably local AngII/Ang1–7 levels and inhibit deleterious RAS effects on remaining at risk tissues in COVID-19 patients.
Conclusion
In the current COVID-19 pandemic, saving lives and increasing hospital capacity for ventilatory support are priorities for many already stretched and often overwhelmed healthcare systems. Currently, we have supportive but very limited therapeutic options for high-risk COVID-19 patients in intensive care settings. In this viewpoint we presented a theoretical framework by which ACE2 replacement or augmentation appears a rational therapeutic option for severely ill, high cardiovascular risk, no-other option COVID-19 subjects. If early phase 1/2 ACE2 replacement proves promising, then expedited compassionate off-label use might also be considered. Given that ACE2 is synthesised using recombinant manufacturing, it is likely that a number of pharmaceutical companies would have the capacity to scale up production quickly for global use.
Acknowledgments
Figures created with Biorender.com. The authors thank Mr Jerry Gu for his useful input on figure design.
References
Footnotes
Contributors NC conceived the concept. NC and PMR drafted the manuscript.
Funding This work was supported in part by Science Foundation Ireland (Research Grant SFI/12/RC/2273).
Competing interests None declared.
Patient consent for publication Not required.
Provenance and peer review Not commissioned; externally peer reviewed.