Article Text

Statistics from Altmetric.com
Learning objectives
Become familiar with the spectrum of cardiac involvement in mitochondrial diseases (MDs).
Recognise the clinical features of cardiac involvement in MDs.
Understand the current strategy for management of MDs.
Introduction
Mitochondrial diseases (MDs) include a wide range of clinical entities involving tissues that have high energy requirements such as heart, muscle, kidney and the endocrine system1 (figure 1). Defects in mitochondrial DNA (mtDNA) mutations are the most common cause of MDs in adults.2 ,3 However, the nuclear gene defects are increasingly recognised as a cause of disease.4 Although the true prevalence of cardiac involvement in MD is unknown, cardiovascular involvement presents specific clinical issues that require systematic evaluation. In addition, in some MD-related mutations (eg, m.3243A.G) cardiac disease is the most common cause of early death.5 Hence, cardiologists are likely to become increasingly involved in the multidisciplinary care of patients with MD.

Clinical features of mitochondrial disease.
Genetics of mitochondria and MD
A detailed description of the genetics of the mitochondria is beyond the scope of this review, but the general principles are summarised in box 1.
Biology and genetics of mitochondria
Structure and function
Mitochondria have two genomes: the mitochondrial (mtDNA) and the nuclear genome (nDNA).
mtDNA contains 37 genes, 28 on the H-strand and 9 on the L-strand. Thirteen of the genes encode one polypeptide component of the mitochondrial respiratory chain (RC).
nDNA encodes components of oxidative phosphorylation system, and over 35 proteins required for the RC assembly (complex I=11 nDNA assembly factors, complex III=2 assembly factors, complex IV=18 assembly factors and complex V=4 assembly factors).
Inheritance
mtDNA is maternally inherited
nDNA inheritance pattern is Mendelian (dominant and recessive)
Homoplasmy (copies of mtDNA are all identical)
Cells contain thousands of molecules of mtDNA.
In the majority of cases their sequence is identical
Heteroplasmy (multiple mtDNA variants in a single cells)
Mutations often co-exist with their wild-type counterpart in various proportion
The mitochondria in the zygote are derived only from the ovum, since the sperm loses the mitochondria necessary to provide movement after fertilisation. This results in non-Mendelian transmission (maternal) from the mother to male and female progeny with typically unpredictable disease penetrance.6 Clinical expression relates to the type of mutation and additional genetic, cellular and environmental modifiers.
However, when the mutation concerns a mitochondrial protein encoded by the nuclear DNA (nDNA), classic Mendelian inheritance pattern is observed.7 Many mutations have been described for the mitochondrial genome and they include over 100 point mutations and 200 deletions. Nevertheless, the number is continuously increasing and duplications and rearrangements have been also described.8 ,9
Mitochondria and the heart
Because the heart is highly dependent on mitochondrial oxidative energy, it is understandable that defects in mitochondrial structure and function can be found in association with different cardiovascular diseases.10 Abnormalities in the mitochondrial function cause cardiomyopathies, arrhythmias and abnormalities of the conduction system (table 1). Mitochondrial dysfunction is also involved in ischaemia-reperfusion injury, heart failure and ageing,11 but this review specifically focuses on primary mitochondrial dysfunction with cardiovascular involvement.
Cardiac involvement in the main mitochondrial syndromes
Mitochondria and cardiomyopathies
The role of mitochondrial dysfunction in cardiomyopathies is supported by a large amount of published literature.12 Hypertrophic cardiomyopathy (HCM) is the dominant phenotype in MD,13 occurring in up to 40% patients. Sarcomeric protein gene mutations are identified in 40%–60% of patients with HCM, but mtDNA-related cardiomyopathy is a rare phenocopy.14 Cardiologists should be aware of this possibility and should be familiar with diagnostic ‘red flags’ for mitochondrial cardiomyopathies15 (box 2).
Common red flags for mitochondrial diseases
Pattern of inheritance
Matrilinear
Systemic sign and symptom
Learning difficulties
Sensorineural deafness
Visual impairment
Muscle weakness
Palpebral ptosis
Electrocardiography
Short PR/pre-excitation
Atrioventricular block
Laboratory tests
Creatine kinase
Transaminase
Lactic acidosis
Myoglobinuria
Leucocytopenia (TAZ gene/Barth syndrome)
Point mutations in mtDNA can cause sporadic or maternally inherited cardiomyopathy, which may be the only or presenting feature of the disease.16 Cohort studies using echocardiography have identified left ventricular hypertrophy in 38%–56% patients harbouring the m.3243A.G mutation and have revealed a correlation between skeletal muscle mutant load and indexed left ventricular mass.17 Patients with high mutation load may therefore be at risk of developing cardiomyopathy. In addition, left ventricular hypertrophy is recognised in patients with other mtDNA mutations including several mt-tRNA genes.18 There are important differences in the cardiac phenotype and natural history of sarcomeric HCM and mtDNA-related cardiomyopathy. Generally, in mitochondrial cardiomyopathies left ventricular outflow tract obstruction is not present and evolution to left ventricular systolic dysfunction is more common than in sarcomeric HCM.19 ,20
Dilated cardiomyopathies presenting phenotype is less common than HCM. Echocardiographic studies in adults suggest that dilated cardiomyopathy may be slowly progressive and, at least in some patients, is responsive to conventional heart failure therapies,21 although data in literature are limited to be conclusive. The association between left ventricular non-compaction and MDs is relatively common in children.22 Abnormalities of mitochondrial function have been described in Barth syndrome, characterised by left ventricular non-compaction, cyclic neutropaenia, hypocholesterolaemia and possible evolution in dilated/hypokinetic phenotype.23 Also, left ventricular hypertrabeculation aspect without non-compaction cardiomyopathy diagnostic criteria associated with progressive systolic impairment may be frequently associated with MDs.24
Mitochondria and arrhythmias
Over the last 20 years, a growing body of evidence indicates that cardiac mitochondria are involved in the genesis of arrhythmias.25 Ventricular pre-excitation and Wolff-Parkinson-White (WPW) syndrome may be more common in patients with mtDNA mutations than in the general population. First observed in association with Leber's hereditary optic neuropathy, ventricular pre-excitation has been reported in 10% patients and 8% maternal relatives.26 ,27 In a literature review of reported cases of mitochondrial encephalomyopathy, lactic acidosis and stroke-like episodes syndrome (MELAS), 6 of 43 patients (14%) had WPW;28 these data have been confirmed in another recent survey.29 The prevalence of WPW is also increased (up to 15%) in patients with myoclonic epilepsy with ragged red fibres (MERRF) syndrome.30
Although ventricular pre-excitation has been reported in association with mtDNA-related cardiomyopathy,31 this combination does not appear as common as in other forms of inherited disease such as that caused by PRKAG2 gene mutations.32
There are few data on the prevalence of arrhythmias in patients with MD. Sudden death is a rare event, but can occur particularly in severe childhood forms,33 or in asymptomatic adults carrying the m.3243A>G mutation, even with no evidence of cardiomyopathy by imaging techniques.34
Mitochondria and cardiac conduction disorders
Conduction disorders are the main cardiac signs encountered in Kearns-Sayre syndrome (KSS) and chronic progressive external ophthalmoplegia syndromes.35 Abnormalities range from simple PR interval prolongation to infranodal high-degree atrioventricular (AV) block. Although mechanisms are currently unknown, differences in mutation load or in sensitivity of different cardiac cell types to mtDNA dysfunction may account for this phenotypic heterogeneity.36 Of note, in patients with neuromuscular disease, progression to high-grade AV block is often unpredictable necessitating prompt recognition of any conduction system disease and consideration of early intervention.37 Risks of progression and clinical outcomes associated with conduction system disease in other forms of mtDNA disease are unknown.
Diagnosis
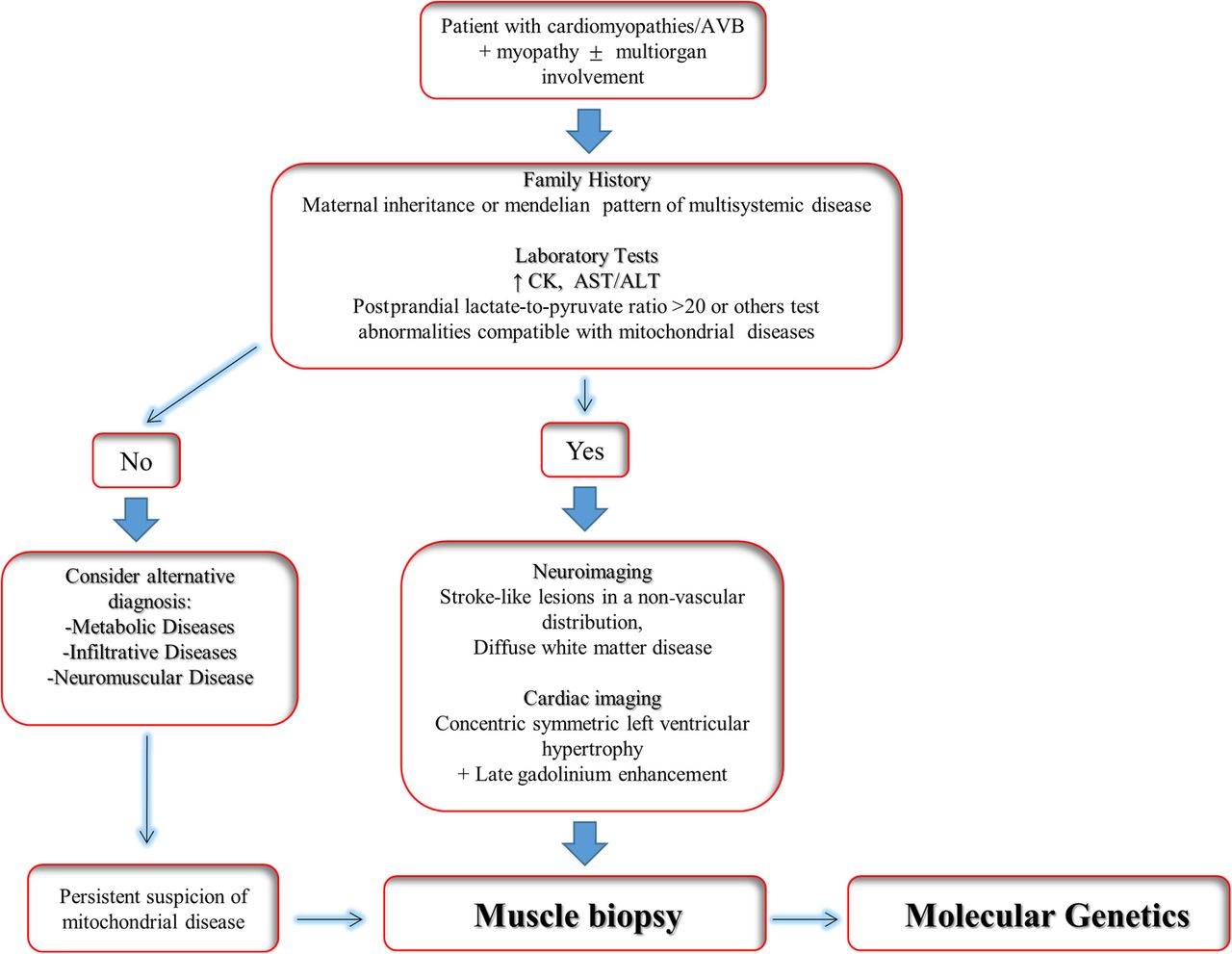
The diagnosis of MD is based on clinical, biochemical, histological/histochemical and genetic criteria.38 These criteria have improved the clinical detection of MD, However, due to the extreme variability in the clinical presentation of MD, a multiparametric approach is required to make the diagnosis (figure 2).
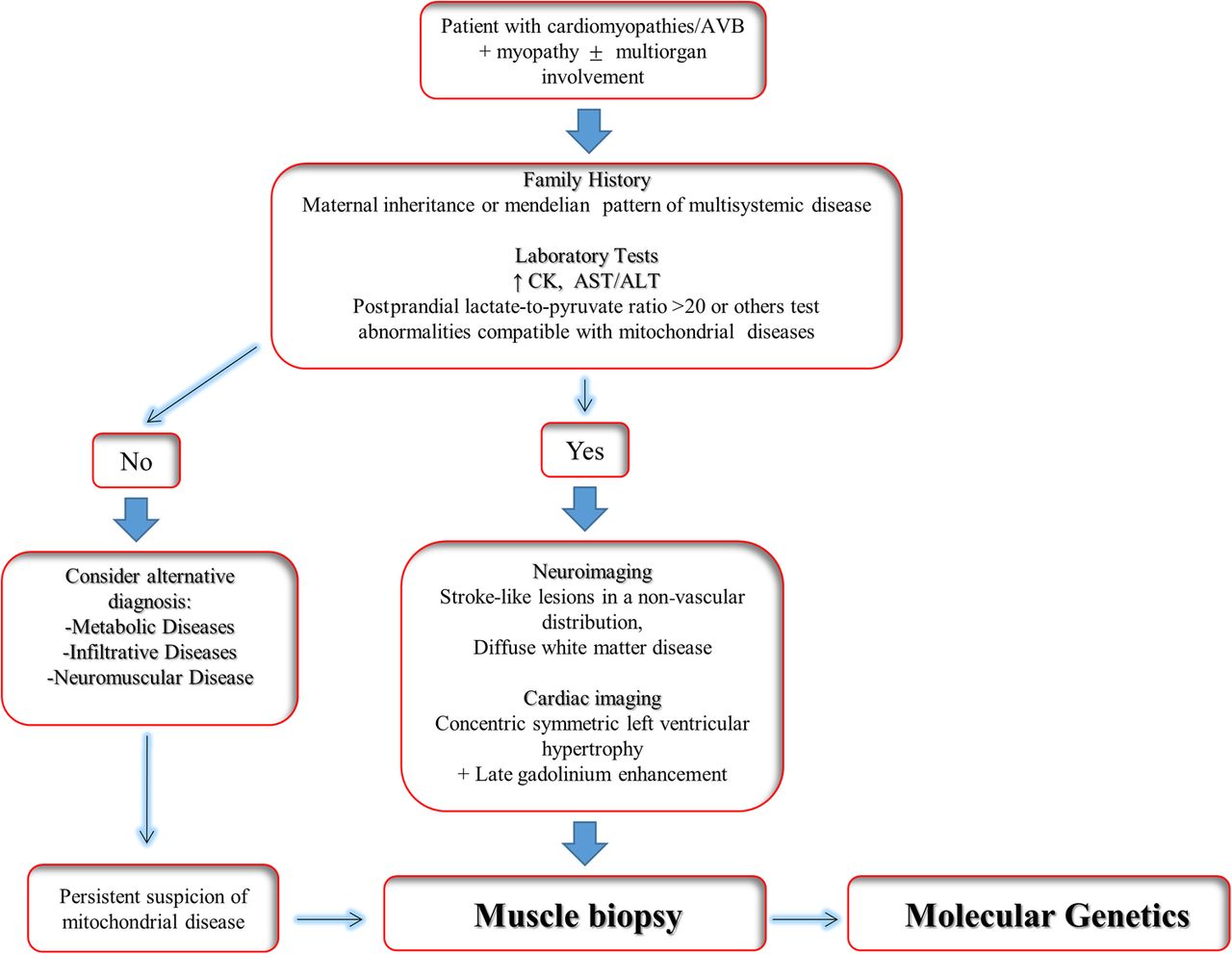
Clinical algorithm for diagnosis of mitochondrial disease. AST/ALT, aspartate transaminase/alanine transaminase; AVB, atrioventricular block; CM, cardiomyopathies; CK, creatine kinase.
Cardiologists managing patients with MD should be aware of the different aspects of the disease, and ideally should link to a specialist team comprising neurologists, geneticists and pathologists. The clinical investigation of a patient with suspected MD should start with a three-generation pedigree in order to detect a family history of maternal inheritance or multisystem disease. A classic Mendelian inheritance pattern is consistent with a nuclear mutation.39 Early onset diabetes, deafness, muscle myopathy, ptosis, alopecia and other signs and symptoms in association with cardiomyopathies and/or arrhythmias in other family members can be suggestive of the diagnosis.15
Laboratory tests
The initial laboratory evaluation for suspected MD should include complete blood count, creatine kinase, transaminases, albumin, lactate and pyruvate, amino acids and acylcarnitines, along with quantitative or qualitative urinary organic acids.40 However, interpretation of these tests requires specific competence in mitochondrial medicine. Thus, for general cardiologists or practitioner the most useful and readily available laboratory tests for suspected MD is serum level of lactate. An increased (>20) postprandial lactate-to-pyruvate ratio is frequent in MD, but a normal ratio does not exclude the disease.41
Electrocardiogram
An early ECG sign of MD is T-wave abnormalities (eg, early phase of MELAS/MERRF),30 even in absence of left ventricular hypertrophy. The presence of a short PR (figure 3A) or WPW, and the presence of various degrees of AV blocks are also supportive evidence of the diagnosis.15

Cardiac features of mitochondrial diseases. (A) ECG of patients with mitochondrial encephalomyopathy with lactic acidosis and ‘stroke-like’ episodes (MELAS) syndrome, showing a short PR interval (see arrow). (B) Two-dimensional (2-D) echocardiography image in short-axis view showing concentric left ventricular (LV) hypertrophy in patients with myoclonic epilepsy with ragged red fibres syndrome (see arrow). (C) 2-D echocardiography image in four chamber view showing marked apical hypertrabeculation in patients with MELAS syndrome (see arrow). (D) Cardiac MRI in short-axis LV view in a patient with Kearns-Sayre syndrome showing concentric LV hypertrophy with late gadolinium enhancement in the inferolateral wall (arrow). Courtesy of Dr Santo Dellegrottaglie.
Cardiac imaging
Although all the cardiomyopathy phenotypes may be seen, the association between left ventricular hypertrophy (with or without apical trabeculation; figure 3B, C), with systolic dysfunction has been reported as typical presentation of evolution of mitochondrial cardiomyopathy.42 ,43
Cardiovascular magnetic resonance (CMR) is a highly sensitive tool for depicting myocardial abnormalities in patients with MD including tissue damage by late gadolinium enhancement (LGE).44 Recently, a large-sized CMR study suggested that characteristic patterns of cardiac involvement might be present in some MD.45
For instance, concentric remodelling or left ventricular hypertrophy with intramural LGE in the inferolateral wall seems typical in patients with KSS (figure 3D).
Neuroimaging in the form of CT and MRI of the brain have been used to assist in the diagnosis of MD. Stroke-like lesions in a non-vascular distribution, and diffuse white matter disease, are known classic findings in syndromic MD.46 ,47 These ‘classical’ changes are selectively also observed in non-syndromic MDs and other metabolic disorders as well.48 ,49 Thus, they are neither sensitive nor specific enough to allow for a primary MD diagnosis without the presence of other abnormalities.
Cardiopulmonary exercise testing
Metabolic stress test (cardiopulmonary) can be of help in mitochondrial myopathies.50 The key features of a mitochondrial myopathy are low anaerobic threshold (indicating impaired or inefficient oxygen utilisation) and increased respiratory exchange ratio, indicating an inefficient utilisation of fatty acids as an energy source.51
Muscle biopsy
Muscle biopsy specimen remains the gold standard for MD diagnosis, even if the specificity and sensitivity is not 100% and the technique is performed in only a few specialised centres. Muscle biopsy (preferably an open biopsy of the vastus lateralis) specimens is routinely examined for structural changes with the use of light microscopy, histochemistry for specific enzymes and ultrastructural examination by electron microscopy.7 Ragged red fibres caused by the subsarcolemmal accumulation of mitochondria as seen on a modified Gomori trichrome stain are considered to be the histological hallmark of MD, but they can be absent in children and are seen only in late-stage disease in many adults.
Genetic testing
Screening of mtDNA can be done for the most common mutations by Sanger sequencing and next-generation sequencing (NGS). Since mtDNA is highly polymorphic, a mutation can be considered pathogenic when segregates with the phenotype in affected families, it is absent in healthy controls and it has been potentially associated with a pathogenetic mechanism by molecular modelling or functional studies.52 Some preliminary experience shows that exome sequencing enhances the ability to identify potential nuclear gene mutations in patients with biochemically defined defects affecting multiple mitochondrial respiratory chain complexes.53 ,54 The association between genotype and phenotype is still under investigation, but identification of causative mutations facilitates presymptomatic diagnosis of family members, allows efficient follow-up and aids preconception genetic counselling.
Therapy
At present, there is no cure for MD. Treatment of MD is aimed at maintaining optimal health, using preventive measures to mitigate symptoms and avoid mitochondrion toxic drugs. Because patients with MD have altered nutritional needs compared with the general population, first-line therapy is based on optimising the nutritional regimen. Evaluation of resting metabolic rate may aid in establishing the ideal caloric intake needed for the patient. Although specific dietary restrictions or changes are not universally recommended, a comprehensive evaluation of a patient’s nutrition and potential deficiencies is needed. Some patients may need restriction of carbohydrate, protein or fat. Caloric supplementation, enteral feeding, limited fasting, increased meal frequency and intravenous nutrition are all potential therapeutic avenues to consider. Treating swallowing dysfunction, abnormal gut motility, behavioural feeding issues and gastro-oesophageal reflux is recommended to allow for optimal nutritional intake.55 Exercise may benefit patients with MD preventing deconditioning, which exacerbates pre-existing exercise intolerance and levels of fatigue and improves muscle oxidative metabolism. It also facilitates decreased lactate levels and an advantageous ATP production.56 Endurance training seems to improve oxygen utilisation and extraction, peak work capacity and submaximal exercise tolerance.57 Endurance training can also induce satellite cell activation and an improvement in biochemical activity of striated muscle.58
According to recent guidelines of the Mitochondrial Medicine Society,59 nutritional supplement with ubidecarenone (coenzyme Q10), α-lipoic acid and riboflavin should be used in all patients with a diagnosis of MD. Folinic acid should be considered in patients with central nervous system manifestations and l-carnitine should be administered only in patients with a documented deficiency.
Patients with MD should avoid certain medications that interfere with mitochondrial function and can precipitate acute or subacute multiorgan failure secondary to worsening mitochondrial respiratory chain function (mitochondrial crisis) like metformin, valproic acid, statins and propofol.
Cardiac involvement requires careful evaluation and management (figure 4). Asymptomatic individuals with pre-excitation do not require active intervention, but an electrophysiological study may be reasonable in some patients (ie, patients with family history of sudden death) to stratify risk of subsequent ventricular fibrillation and those that may become symptomatic later in life.60

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Algorithm for clinical management of mitochondrial disease. ESC, European Society of Cardiology; HCM, hypertrophic cardiomyopathy; HF, heart failure; SD, sudden death.
Conversely, patients with WPW and symptomatic tachycardia will require radio frequency catheter ablation.61 Moreover, catheter ablation is also indicated in asymptomatic patients with accessory pathways capable of rapid antegrade conduction (effective refractory period <240 ms) due to high risk of sudden death if atrial fibrillation occurs.62
In many patients with MD, second-degree AV block remains stable over decades. However, progressive heart block and sudden death are recognised. The American College of Cardiology/American Heart Association/Heart Rhythm Society 2012 Guidelines for device-based therapy of cardiac rhythm abnormalities recommend permanent pacemaker implantation for third-degree and advanced second-degree AV block at any anatomic level associated with neuromuscular diseases with AV block, including KSS, with or without symptoms.63 Given the unpredictable nature of progression of heart conduction defects in MD and neuromuscular disorders, a permanent pacemaker implantation may be considered for patients with any degree of AV block (including first-degree AV block with PR >300 ms), or any fascicular block with or without symptoms, especially if there is evidence of progressive abnormalities.
Management of supraventricular tachyarrhythmias in patients with MD (especially atrial fibrillation) must be conducted according to international guidelines.64 With few exceptions, there is no current evidence that patients with MD have an increased risk of life-threatening ventricular arrhythmia, in the absence of significant left ventricular dysfunction or conduction system disease. Thus, risk stratification for sudden death should follow current clinical practice guidelines.65 One important exception is patients with m.3243A>G, in which sudden death can occurs also in absence of myocardial involvement. Indeed, a recent study suggests that sudden death is an unrecognised clinical entity in young, asymptomatic adults with m.3243A.G disease.34 In these patients, unexplained syncope or a family history of sudden death should prompt the use of loop recorder. Implantable cardioverter defibrillator is reasonable when episodes of non-sustained ventricular tachycardia occur.34
In some patients, inappropriate sinus tachycardia due to persistent lactic acidosis or to dysautonomia may occur. In these patients, use of β-blockers or ivabradine may be useful. In all patients with evidence of myocardial involvement, conventional therapy with β-blockers (in the absence of AV block), ACE inhibitors or angiotensin receptor blockers should be initiated, regardless of symptomatic status.
In the end-stage phases of mitochondrial cardiomyopathy cardiac transplant may be considered. However, transplantation is controversial since multiorgan involvement is usually accepted as a contraindication. Neuromuscular weakness can present difficulties during and after anaesthesia and the prolongation of life may potentially lead to long-term neurological disability. According to National Institute for Health and Care Excellence, cardiac transplantation is reasonable in isolated mitochondrial cardiomyopathy66 and successful orthotopic cardiac transplantations have been reported in such cases.67
Clinical course in children and adults
The natural history of MD differs between children and adults. In the paediatric age group, clinical prognosis is strictly related to cardiac involvement.68 In a large retrospective study, Scaglia et al showed that patients with MD and cardiomyopathy had an 18% survival rate at 16 years of age, instead patients with exclusive neuromuscular features but no cardiomyopathy had a 95% survival at the same age. The prevalence and natural history of cardiovascular disease in adult patients with MD is less well characterised. However, despite cardiac involvement is frequent, cardiovascular complications rarely occur, and the prognosis is generally favourable.7 ,30–69 An exception is represented by m.3243A>G patients in which life-threatening events can occur.5 ,7 ,34–69
Conclusion
MD represent a heterogeneous group of inherited disorders characterised by defects in the mtDNA and nDNA, leading to a spectrum of clinical entities.70 Because diagnosis is a challenge for any clinician, the first rule is maintaining a high degree of suspicion. Patients should be referred to physicians with the appropriate expertise in MD whenever the diagnosis is suspected. Patients with cardiac involvement should be counselled about cardiovascular complications and management. Current pharmacological strategies are incompletely effective, and large randomised controlled trials are required to direct future therapy.
Key messages
Mitochondrial disease (MDs) can occur at any age with a wide range of cardiac and non-cardiac symptoms.
Hypertrophic cardiomyopathy is the most frequent manifestation of MDs. However, all forms of cardiomyopathy have been described. Wolff-Parkinson-White (WPW) and ventricular arrhythmias are part of the clinical spectrum.
Clinical diagnosis of MD requires a comprehensive, multispecialty approach. In familial diseases (cardiomyopathies and/or arrhythmias), the presence of a wide array of symptoms (‘red flags’), including muscle weakness, diabetes, deafness, should raise the suspicion of a mitochondrial disorder. A matrilinear inheritance (mitochondrial DNA-related diseases) is highly suggestive of mitochondrial disorders. ECG (atrioventricular (AV) blocks and/or short PR) and laboratory tests (increased creatine kinase, transaminases, lactate/pyruvate ratio) represent useful tools for the diagnosis.
The prevalence and natural history of cardiovascular disease in MD is poorly characterised. However, recent studies report a relatively high frequency of life-threatening events in patients with m.3243A>G mutation.
There is no cure for mitochondrial disorders. In all patients with evidence of myocardial involvement, conventional therapy with β-blockers (in the absence of AV block), ACE inhibitors or angiotensin receptor blockers should be initiated, regardless of symptomatic status. In the end-stage phases of mitochondrial cardiomyopathy, cardiac transplant may be considered. However, transplantation is controversial since multiorgan involvement is usually accepted as a contraindication.
You can get CPD/CME credits for Education in Heart
Education in Heart articles are accredited by both the UK Royal College of Physicians (London) and the European Board for Accreditation in Cardiology—you need to answer the accompanying multiple choice questions (MCQs). To access the questions, click on BMJ Learning: Take this module on BMJ Learning from the content box at the top right and bottom left of the online article. For more information please go to: http://heart.bmj.com/misc/education.dtl
RCP credits: Log your activity in your CPD diary online (http://www.rcplondon.ac.uk/members/CPDdiary/index.asp)—pass mark is 80%.
EBAC credits: Print out and retain the BMJ Learning certificate once you have completed the MCQs—pass mark is 60%. EBAC/ EACCME Credits can now be converted to AMA PRA Category 1 CME Credits and are recognised by all National Accreditation Authorities in Europe (http://www.ebac-cme.org/newsite/?hit=men02).
Please note: The MCQs are hosted on BMJ Learning—the best available learning website for medical professionals from the BMJ Group. If prompted, subscribers must sign into Heart with their journal's username and password. All users must also complete a one-time registration on BMJ Learning and subsequently log in (with a BMJ Learning username and password) on every visit.
References
Footnotes
Contributors GL and DM: writing the article, critical revision of the article, final approval of the article. GP: critical revision of the article, final approval of the article.
Competing interests None declared.
Provenance and peer review Commissioned; externally peer reviewed.