Article Text

Statistics from Altmetric.com
While mortality from overt ischaemic heart disease is falling, that from heart failure is increasing and reaching epidemic proportions.1 Although an aging population is partly responsible for this trend, recent observations showing persistently high mortality and morbidity rates following myocardial infarction further compounds the issue.2 An understanding of the pathophysiological processes leading to heart failure, and in particular the mechanisms underlying postinfarction heart failure (postinfarction ventricular remodelling) is therefore fundamental and forms the basis of this review.
Acute myocardial infarction
Immediately after coronary artery occlusion, irreversible cell necrosis can occur within minutes. Factors affecting the amount of necrosis include the presence or absence of a preconditioning stimulus,3 the volume of ischaemic myocardium, and the amount of collateral blood flow.4 In addition, prompt reperfusion within a narrow time window, when myocytes are in a state of critical ischaemia and are prenecrotic/viable, has been shown to reduce cell death, limit infarct size, and increase survival.5
Myocardial infarction is followed by a complex and interrelated sequence of events termed postinfarction left ventricular remodelling (table 1) which describes the compensatory responses of the cardiovascular system when faced with an acute loss of myocardial contractile function.6 Morphologically, the end result of these responses can be quantified by imaging the infarcted ventricle as it progressively dilates and assumes a more spherical as opposed to elliptical contour.6
Components of postinfarction ventricular remodelling
In the early stages, ventricular dilatation can be considered beneficial as it maintains stroke volume through the Frank-Starling mechanism. At the same time, however, the dilating ventricle proves detrimental as it exerts further demands on the surviving myocardium through increases in wall tension (the law of Laplace, fig1).7 Long term, left ventricular volume is a sensitive marker of postinfarction ventricular dysfunction, left ventricular end systolic volume being one of the most powerful independent predictors of prognosis after myocardial infarction.8
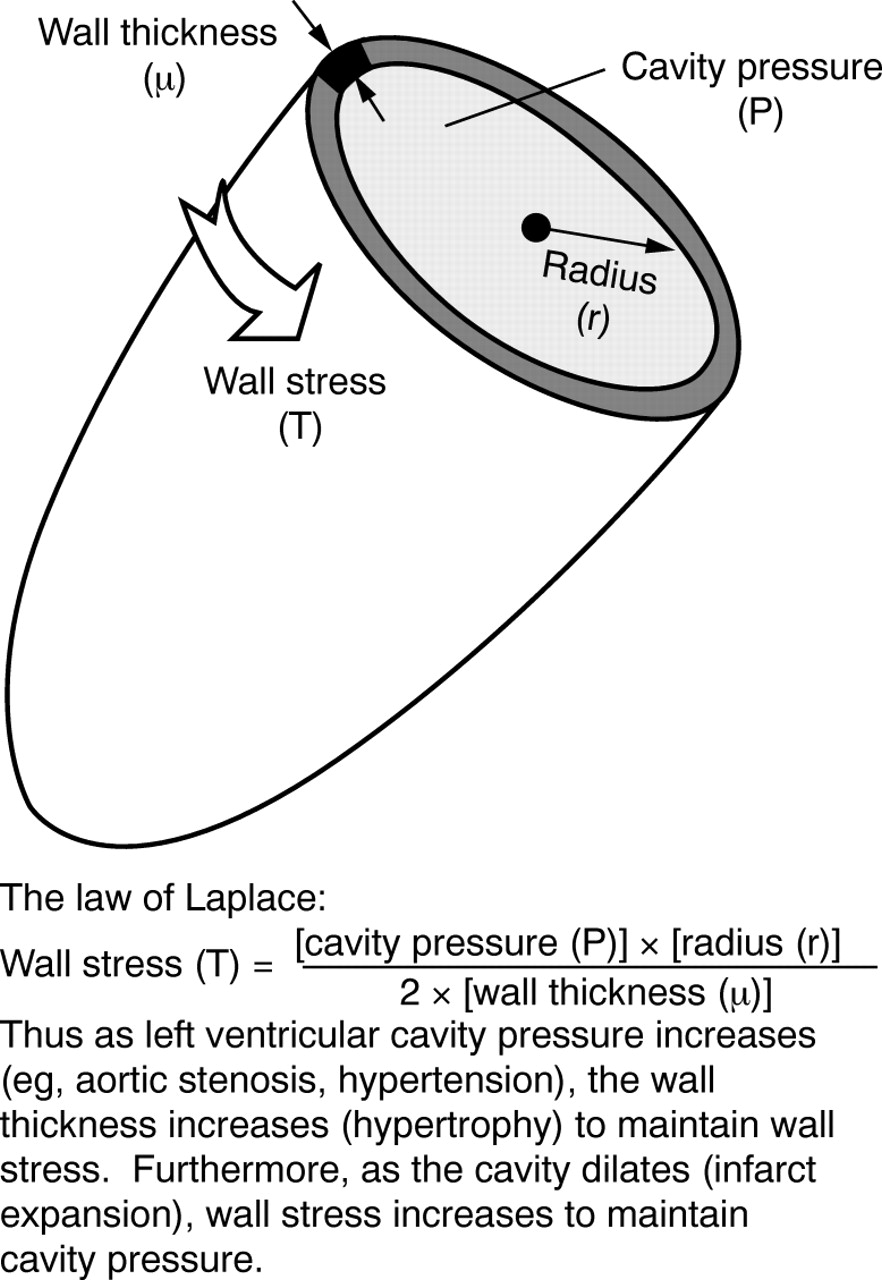
The law of Laplace.
Infarct expansion
Cell necrosis initiates an inflammatory reaction with granulocyte infiltration and release of proteolytic enzymes. The matrix metalloproteinases (MMP), a family of zinc dependent collagenases and their endogenous inhibitors (tissue inhibitors of MMP, TIMP) play a crucial role in subsequent collagen degradation and infarct expansion.9 Collagen breakdown predominates up to 14 days following infarction when serum levels of MMP-1 are raised. Thereafter, serum TIMP-1 concentrations prevail and activity shifts from lysis to fibroblast infiltration, collagen deposition, and scar formation.10
During the early lytic phase, the infarcted segment is vulnerable to pressure and shape change within the ventricle which increase wall stress and strain and result in lengthening and thinning through lateral slippage of myocardial planes.11 The surface area occupied by the infarcted region thus increases and the infarct zone expands. The magnitude of infarct expansion depends on various factors (table 2), which, when extreme, can lead to left ventricular or septal rupture.
Factors associated with infarct expansion
Neurohormonal activation
Plasma concentrations of several neurohormones are increased after myocardial infarction. The magnitude and time course of activation are closely related to infarct size and degree of left ventricular dysfunction.12 The neurohormonal hypothesis postulates that neurohormones initially serve an adaptive role by maintaining cardiac output, but in the later stages the responses becomes pathological and contribute to adverse remodelling, progressive ventricular failure, and ultimately to the syndrome of heart failure.13
AUTONOMIC SYSTEM
One of the earliest responses during infarction is an increase in sympathetic drive which has both positive chronotropic and inotropic actions. While cardiac output is maintained, left ventricular loading increases, thereby aggravating infarct expansion and promoting adverse ventricular remodelling. In addition, myocardial oxygen demand rises, precipitating further ischaemia, and the proarrhythmic effects of catecholamines may predispose to fatal arrhythmias.
In the long term, sympathetic activation contributes to the re-expression of a fetal gene program and ventricular hypertrophy which may ultimately exacerbate ventricular dilatation.14 The corollary that sympathetic antagonists confer advantage has been shown in several randomised β blocker trials including a meta-analysis which concluded that treatment with β adrenoceptor blocking drugs for 12 months after myocardial infarction reduces mortality by up to 25%.15 More recently, vasodilating β blockers have been shown to reduce the risk of all cause mortality in established heart failure by up to 65% after just six months of treatment,16 while in another similar randomised, double blind, placebo controlled study, the addition of a conventional β blocker (bisoprolol) to “standard” medical treatment resulted in a comparable reduction in all cause mortality (34%) following 18 months of treatment.17
THE RENIN-ANGIOTENSIN AXIS
Vasoconstriction and fluid retention are both actions of the renin-angiotensin axis which serve to maintain blood pressure. In common with catecholamines, left ventricular afterload eventually rises and adverse remodelling is promoted. A reduction in the activity of this axis with angiotensin converting enzyme (ACE) inhibitors thus appears appropriate and forms the rationale for the large ACE inhibitor trials. Such mechanisms are likely to underlie the 11% reduction in mortality after myocardial infarction observed at six weeks in the GISSI-3 trial (Gruppo Italiano per lo Studio della Sopravvivenza Nell'Infarto Miocardico), where ACE inhibitors were used in the first 24 hours of infarction.18 Similar processes may explain the late benefits of ACE inhibitors after infarction. For example, a 21% risk reduction of cardiovascular mortality was observed after an average follow up of 42 months in patients treated with captopril (started three to 16 days after myocardial infarction) compared with placebo matched controls.19 Comparable benefits of enalapril are also seen in patients with established left ventricular dysfunction (but not necessarily a history of myocardial infarction), with a 22% reduction in death from progressive heart failure after a mean follow up of 41 months.20 These effects might reflect a reduction of systemic angiotensin II, which, in addition to its hypertensive effects, has various other detrimental myocardial actions.21
More recently, antagonists of the angiotensin II type 1 receptor (AT1-R) have been developed. The ELITE study (evaluation of losartan in the elderly) compared captopril (an ACE inhibitor) with losartan (an AT1-R antagonist) in elderly, ACE-I naive patients.22Although the trial was designed to examine a primary end point of renal tolerability, a trend towards a reduction in all cause mortality in the losartan group was observed (4.8% v 8.7% after 48 weeks of treatment).22 ELITE II, an ongoing trial designed to examine all cause mortality, may further our understanding of the mechanisms underlying postinfarction ventricular remodelling including the relative actions played by the kinins, for example bradykinin.
NATRIURETIC PEPTIDES
The three principal members of the natriuretic peptide family (atrial natriuretic factor, ANF; brain natriuretic peptide, BNP; and C-terminal of atrial natriuretic peptide, CNP) exert their phenotypic effects through two main receptors (A and B). Systemic levels of the peptides are increased following myocardial infarction, with ANF being released from the right atrium in response to increases in right atrial stretch, and BNP (and to a lesser degree CNP) being released from the ventricles in response to raised right ventricular wall tension.23 ANF and BNP exert opposite effects to the sympathetic outflow and the renin-angiotensin axis by reducing afterload through a combination of peripheral vasodilatation and natiuresis.23 In addition to their early beneficial effects on haemodynamics, fluid balance, and renal function, there is experimental evidence that the natriuretic peptides may in the long term inhibit myocyte hypertrophy and therefore foster an environment for beneficial remodelling.24
However, their use clinically has so far been unsuccessful. The peptides have a short half life, and tolerance to their effects develops rapidly. Furthermore, potent hypotension and bradycardia have limited their use, though novel approaches involving TIMP inhibiting natriuretic peptide clearance are underway in experimental models. In a recent study, for example, cardiomyopathic hamsters treated with omapatrilat (a unique TIMP with combined neutral endopeptidase and angiotensin converting enzyme inhibitor properties) survived significantly longer than similar captopril treated hamsters.25
ENDOTHELIN
The endothelins (ET-1, ET-2, and ET-3) are potent vasoconstrictor and hypertrophic peptides which are released from vascular endothelium. Systemic concentrations are raised following infarction and contribute to peripheral vasoconstriction, thereby maintaining blood pressure.26 Their effects are similar to the catecholamines and angiotensin II: initially blood pressure is maintained at the expense of increased afterload, and later their hypertrophic effects maintain ventricular function until the balance is tipped, leading to adverse remodelling and progressive ventricular dilatation.27
Antagonists to the two main endothelin receptors, A and B, have been used in animal studies. In a postinfarction rat model of heart failure, long term treatment with bosentan (a combined A and B receptor antagonist) resulted in benefical remodelling compared with placebo matched controls.28 In addition to a significant survival benefit in the bosentan group (65% v 47%), active treatment also resulted in improved haemodynamic stability, a reduction in plasma catecholamines, and reduced left ventricular collagen density (myocardial fibrosis).28 Furthermore, ex vivo analysis after two months of treatment showed a reduction in left ventricular dilatation and hypertrophy.28
Human studies with bosentan, however, are limited through concerns over safety. For example, the REACH-1 trial (research on endothelin antagonism in chronic heart failure) was terminated prematurely when an unprecedented proportion of patients receiving bosentan developed raised plasma transaminases.29 Although only 47% of patients completed the study, bosentan treatment resulted in a significant clinical improvement, a reduction in symptomatic deterioration, and a reduction in the total number of hospital admissions by 40%.29
Thus although interruption of the endothelin pathway is an attractive target with potential advantages, long term pharmacological studies with non-toxic agents are needed before phase III trials can occur in patients with cardiac failure and hypertension.
Myocardial hypertrophy
Adult myocytes are highly specialised terminally differentiated cells and as such have probably lost the ability to divide. Their response to an increased workload is an increase in size and protein synthesis, in addition to an increased production and assembly of sarcoplasmic contractile units.30 The hypertrophic stimuli consist of neurohormonal agents (for example, noradrenaline, angiotensin II, endothelin), local growth promoting peptides (insulin-like growth factor I, cardiotrophin I, fibroblast growth factor), and physical factors causing myocyte stretch (increased preload and afterload, increased wall tension).14 The stimuli act on specific cell surface receptors which activate a cascade of intracellular signalling pathways (fig 2).

{kind=link}
{kind=link}
Signalling pathways associated with myocardial hypertrophy. A simplified overview of our current understanding of the intracellular signalling pathways leading to myocardial hypertrophy. Although the molecular events coupling membrane receptors to nuclear events have not been fully documented, three principal arms (with interpathway crosstalk) are described, each converging onto a common pathway (mitogen activated protein kinase cascade) which ultimately leads to the nucleus. Considerable work remains to be done to fully characterise the pathways before any potential therapeutic targets can be identified.
Minutes after membrane receptor activation, induction of early response genes (for example, c-fos, c-jun, and c-myc) may be observed.31 These genes (also termed proto-oncogenes) are functionally analogous to virus associated oncogenes and are responsible for the synthesis of small regulatory proteins which control the transcription of other genes. There follows a re-expression of a fetal gene program, which in animal models includes the induction of contractile proteins (β myosin heavy chain, α skeletal actin, and β tropomyosin), and non-contractile proteins (ANF and β2 Na/K-ATPase) which ordinarily are only detectable in the fetal period when global cellular hyperplasia predominates.32 A similar reversion to the fetal genome may be observed in adult human cells capable of division when stimulated appropriately (for example, the re-expression of α fetoprotein by acutely damaged hepatocytes following infective hepatitis).33 It appears therefore that adult myocytes may experience similar molecular signals to other cells when faced with a noxious stimulus, but whereas other cells are able to respond by mitosis, myocytes which are arrested in the G0 phase of the cell cycle undergo hypertrophy which appears to be the only growth response available.
Global ventricular dilatation
In addition to early left ventricular dilatation (infarct expansion), the ventricle undergoes a later and more insidious process of further dilatation when other compensatory mechanisms fail to maintain cardiac output.6 In general, where the infarcted region occupies over 10% of the total myocardial mass (as occurs in approximately 40% of all transmural anterior myocardial infarctions) the ventricle progressively enlarges, heralding the onset of symptomatic heart failure and its associated hazards.34
Initially, hypertrophy involves the serial deposition of new sarcoplasmic elements, which results in elongation of cells without an increase in cell thickness (eccentric hypertrophy).6 This pattern of hypertrophy causes global left ventricular dilatation and wall thinning which increases wall tension (the law of Laplace).7 In addition, these cardiocytes may have an impaired contractility which is further compounded by the greater left ventricular mass to cavity volume ratio, leading to relative ischaemia as the capillary network cannot keep pace with tissue growth and is unable to support the greater demands of the hypertrophied tissue.6
Ultimately a vicious cycle is established where loss of contractile tissue activates several adaptive mechanisms which, when unable to compensate adequately, lead to persistently abnormal loading conditions. These in turn exaggerate the compensatory responses which have already failed. The earlier beneficial effects now become maladaptive and the abnormal loading conditions are further enhanced, thus fuelling the downward spiral that perpetuates ventricular dysfunction. The end result is a progressive dilatation of the left ventricular leading to symptomatic heart failure and ultimately death.
Intervention and postinfarction ventricular remodelling
The advantages gained from reperfusing viable tissue are undisputed.5 However, the potential benefits of recanalising vessels supplying an infarcted and non-viable region remain controversial.35 In animal models, late reperfusion following experimental myocardial infarction at a point beyond myocardial salvage significantly reduces infarct expansion and benefits remodelling.36 In humans, observational studies in the postinfarction period have consistently shown survival benefits in the presence of a patent infarct related artery.37 Studies randomising patients after myocardial infarction to medical treatment with or without intervention are, however, few and conflicting. In the SWIFT study (Should We Intervene Following Thrombolysis?), patients surviving acute myocardial infarction were randomised to either conservative care or early angiography plus intervention for patients with stenotic but non-occlusive disease of the infarct related artery.38 A trend towards increased death or non-fatal reinfarction one year later was shown in the interventional group (19.1% v 16.6%; odds ratio 1.19).38 In a similar study with over 3000 patients, no differences were observed between patients managed aggressively or conservatively (combined rate of death and non-fatal myocardial infarction 12 months after myocardial infarction 14.7%v 15.2%, respectively).39However, a more recent study randomising 83 patients after anterior myocardial infarction to medical treatment with or without PTCA reported a significant reduction in left ventricular end systolic volume at six months, and increased five year cardiac event-free survival in the PTCA group.40 Thus although interventional strategies aimed at either halting or reversing adverse postinfarction remodelling are attractive, data supporting these approaches remain speculative and we must await the results of ongoing multicentre studies investigating the “open artery hypothesis”35in order to determine whether there is long term benefit in opening occluded coronary arteries beyond the time window of salvage.
Conclusions
The socioeconomic burden of heart failure is projected to increase. Current treatment, which includes ACE inhibitors and β blockers, only partially reduces the high mortality, which is similar five years after presentation to that of many malignancies. An increase in prevalence of heart failure and an increase in diseased rather than disease free survival will combine to increase the burden of this condition on society.
Further interventions aimed at reducing infarct size, supporting the extracellular matrix, modulating neurohormones, and producing regression of maladaptive cellular hypertrophy are needed if we are to improve the long term prognosis and reduce the morbidity of patients after myocardial infarction. In addition, the role of intervention in this setting requires clarification through additional randomised clinical trials.
Acknowledgments
ZY, SRR, and MSM are supported in part by The British Heart Foundation.