Article Text

Statistics from Altmetric.com
- Torsades de pointes
- drug induced QT prolongation
- antiarrhythmics
- antihistamines
- antimicrobials
- antidepressants
In 1966, Francois Dessertenne described a specific electrocardiographic form of polymorphic ventricular tachycardia, which he termed “torsades de pointes” (TdP).w1 w2 The word “torsades” refers to an ornamental motif imitating twisted hairs or threads as seen on classical architectural columns, and “pointes” referred to points or peaks.w1 w2 In the seminal article, Dessertenne made no attempt to suggest the mechanism of TdP and, until recently, there has been considerable conjecture as to the pathophysiology of this arrhythmia.
CAUSES OF TORSADES DE POINTES
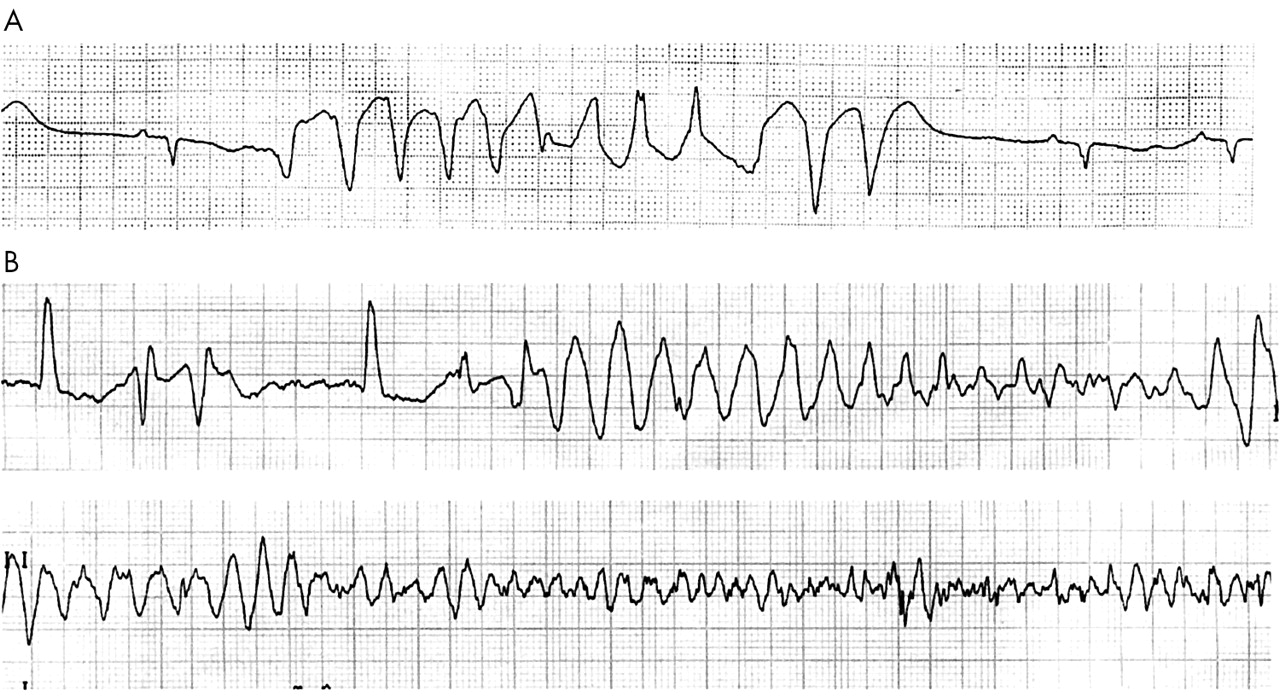
Since the original work by Dessertenne, it has been well recognised that many conditions may cause prolonged or abnormal repolarisation (that is, QT interval prolongation and/or abnormal T or T/U wave morphology), which is associated with TdP. If TdP is rapid or prolonged, it can lead to ventricular fibrillation and sudden cardiac death (fig 1). Essentially, TdP may be caused by either congenital or acquired long QT syndrome (LQTS). In recent years, there has been considerable renewed interest in the assessment and understanding of ventricular repolarisation and TdP. There are several reasons for this. Firstly, the cloning of cardiac ion channels has improved the understanding of the role of ionic channels in mediating cardiac repolarisation, the pathophysiological mechanism of LQTS (congenital and acquired forms), and the pathogenesis of TdP. Secondly, modern molecular techniques have unravelled the mutations in genes encoding cardiac ion channels that cause long QT syndrome, although the genetic defects in about 50% of patients are still unknown. Thirdly, there has been considerable enthusiasm for the development and use of class III antiarrhythmic drugs, which prolong repolarisation and cardiac refractoriness. Unfortunately, drugs that alter repolarisation have now been recognised to increase the propensity for TdP. Finally, an increasing number of drugs, especially non-cardiac drugs, have been recognised to delay cardiac repolarisation and to share the ability with class III antiarrhythmics to cause TdP occasionally.

(A) Self limiting torsades de pointes (TdP). (B) TdP leading to ventricular fibrillation.
Many of the drugs that were initially known to prolong the QT interval were antiarrhythmics, and quinidine was the most commonly implicated agent. Surprisingly, many non-cardiac drugs have also been reported to cause QT prolongation and/or TdP recently. In a survey in both the UK and Italy, non-cardiac drugs that have pro-arrhythmic potential (that is, have an official warning on QT prolongation or TdP, or with published data on QT prolongation, ventricular tachycardia, or class III effect) alone represented 3% and 2% of total prescriptions in both countries, respectively.1 The danger of drug induced pro-arrhythmia is therefore serious. This issue has been identified as a considerable public health problem and has attracted attention from the drug regulatory authorities.
The exact incidence of drug induced TdP in the general population is largely unknown. Most of our understandings of the incidence, risk factors, and drug interaction of pro-arrhythmic drugs are derived form epidemiological studies, anecdotal case reports, clinical studies during drug development, and post-marketing surveillance. The awareness of drug induced TdP in the last few years has resulted in an increase in the number of spontaneous reports. Nevertheless, the absolute total number remains very low, although it has been suggested that the system of spontaneous reporting under-reports the true incidence of serious adverse reactions by a factor of at least 10.2 Between 1983 and December 1999, 761 cases of TdP, of which 34 were fatal, were reported to the World Health Organization Drug Monitoring Centre by the member states.3 The WHO data provide an insight into the incidence of TdP on the most commonly reported pro-arrhythmic drugs3(table 1). However, such a reporting system is undermined by the widely variable content and clinical information between different countries and sources. It is also compounded by various factors such as the patient’s underlying disease, whether the adverse drug reaction is well known or has not been previously described, and the amount of attention paid by the medical community on a specific adverse drug reaction. In this article, we will review the risk of drug induced QT prolongation and/or TdP.
Twenty most commonly reported drugs associated with torsades de pointes (TdP) between 1983 and 19993
MECHANISM OF DRUG INDUCED QT PROLONGATION AND TORSADES DE POINTES
At the cellular level, the repolarisation phase of the myocytes is driven predominantly by outward movement of potassium ions. A variety of different K+ channel subtypes are present in the heart. Two important K+ currents participating in ventricular repolarisation are the subtypes of the delayed rectifier current, IKr (“rapid”) and IKs (“slow”). Blockade of either of these outward potassium currents may prolong the action potential. IKr is the most susceptible to pharmacological influence. It is now understood that virtually without exception, the blockade of IKr current by these drugs is at least in part responsible for their pro-arrhythmic effect. Blockade of the IKr current manifests clinically as a prolonged QT interval and the emergence of other T or U wave abnormalities on the surface ECG. The prolongation of repolarisation may result in subsequent activation of an inward depolarisation current, known as an early after-depolarisation, which may promote triggered activity. When accompanied by the presence of a notably increased dispersion of repolarisation, this may induce re-entry and provoke TdP, which is then sustained by further re-entry or spiral wave activity (fig 2). Such phenomena are more readily induced in the His-Purkinje network and also from a subset of myocardial cells from the mid ventricular myocardium, known as M cells.4 Compared to subendocardial or subepicardial cells, M cells show much more pronounced action potential prolongation in response to IKr blockade.4 This property results in a pronounced dispersion of repolarisation (that is, heterogeneous recovery of excitability), creating a zone of functional refractoriness in the mid myocardial layer, which is probably the basis of the re-entry that is sustaining the TdP.
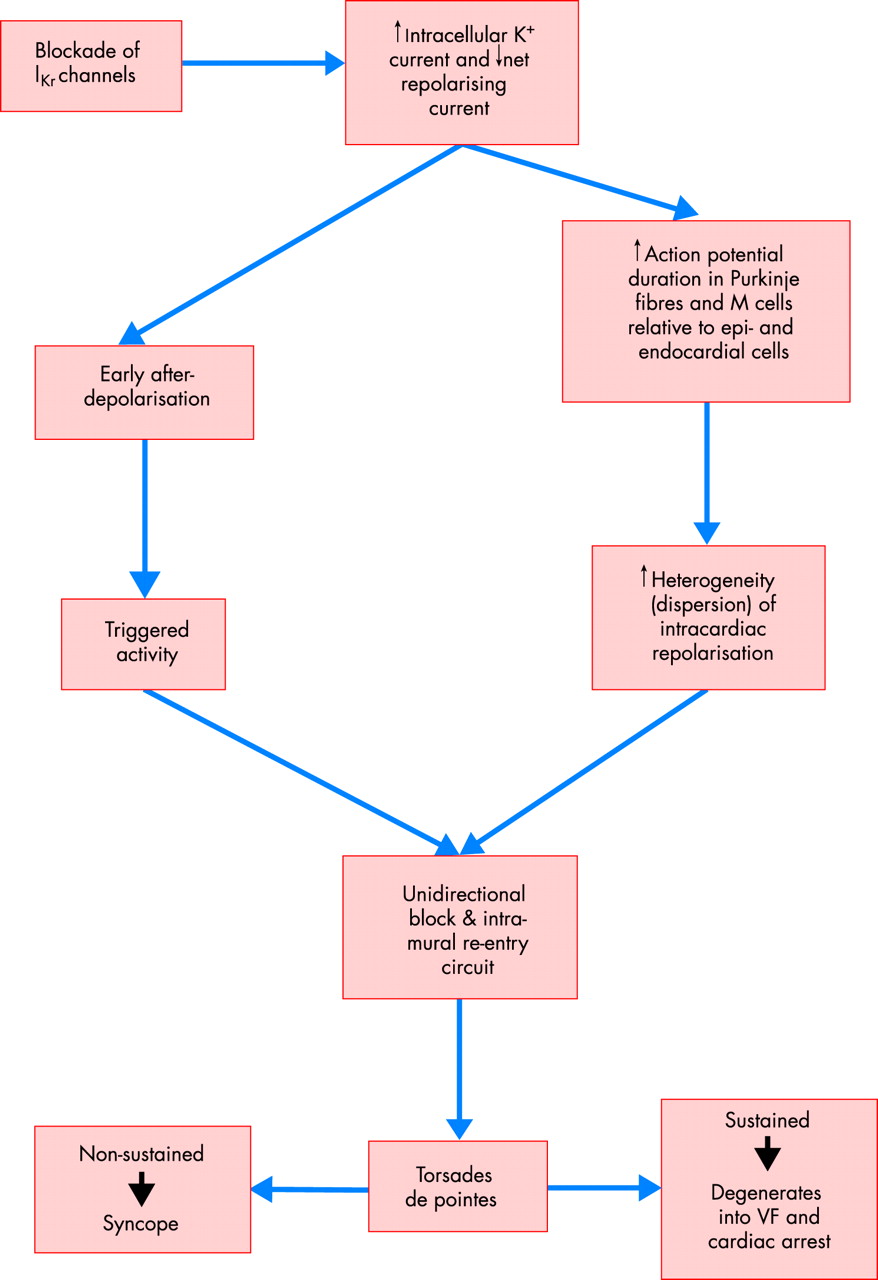
Arrhythmogenesis of torsades de pointes. VF, ventricular fibrillation.
There is a characteristic initiating sequence before the onset of TdP, particularly in the acquired form. The first ventricular complex of the sequence is usually a ventricular ectopic beat or the last beat of a salvo of ventricular premature beats (fig 3). This is then followed by a compensatory pause terminated by a sinus beat. The sinus beat frequently has a very prolonged QT interval and an exaggerated U wave. A ventricular extrasystole then falls on the exaggerated U wave of the sinus beat and precipitates the onset of TdP. It has been suggested that post-pause accentuation of the U wave, if present, may be a better predictor of drug induced TdP than the duration of QTc interval.5
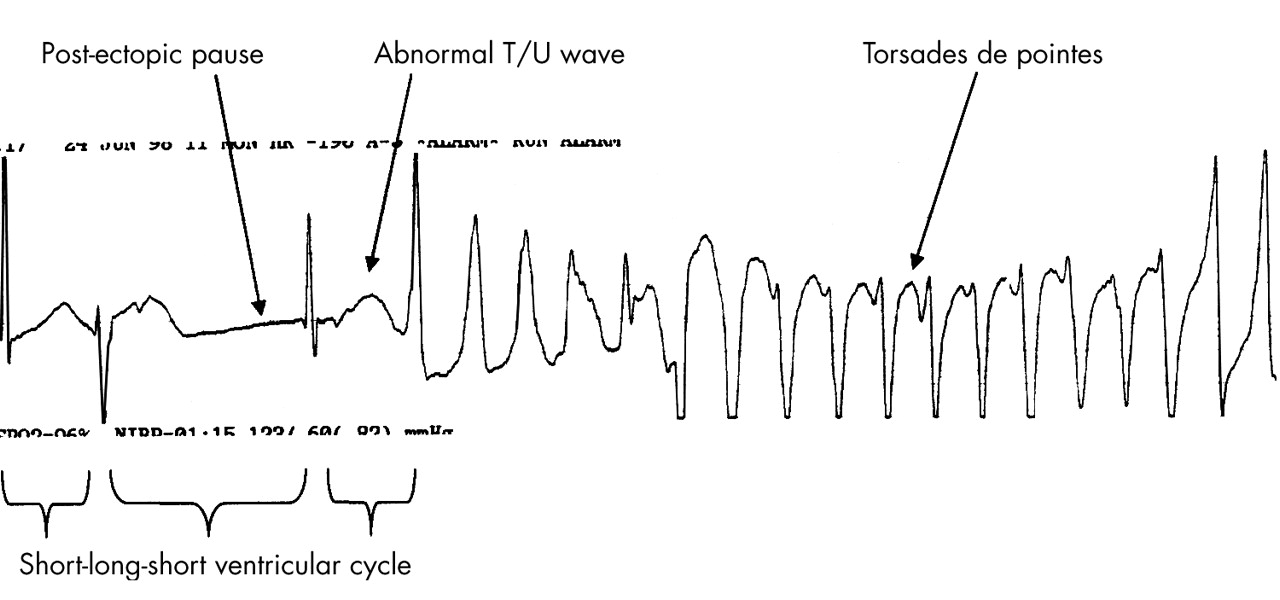
Rhythm strip in a patient with drug induced TdP. Note the typical short-long-short initiating ventricular cycle, pause dependent QT prolongation, and abnormal TU wave leading to the classical “twisting of a point” of the cardiac axis during TdP.
METHOD FOR MEASUREMENT OF QT INTERVAL
When measuring the QT interval, the ECG is best recorded at a paper speed of 50 mm/s and at an amplitude of 0.5 mV/cm using a multichannel recorder capable of simultaneously recording all 12 leads. A tangent line to the steepest part of the descending portion of the T wave is then drawn. The intercept between the tangent line and the isoelectric line is defined as the end of the T wave.w3 The QT interval is measured from the beginning of the QRS complex to the end of the T wave on a standard ECG. There are no available data on which lead or leads to use for QT interval measurement. Traditionally, lead II has been used for QT interval measurement because in this lead, the vectors of repolarisation usually result in a long single wave rather than discrete T and U waves.6
Generally, QT prolongation is considered when the QTc interval is greater than 440 ms (men) and 460 ms (women), although arrhythmias are most often associated with values of 500 ms or more (table 2). The severity of pro-arrhythmia at a given QT interval varies from drug to drug and from patient to patient. Unfortunately, the extent of QT prolongation and risk of TdP with a given drug may not be linearly related to the dose or plasma concentration of the drug because patient and metabolic factors are also important (for example, sex, electrolyte concentrations, etc). Furthermore, there is not a simple relation between the degree of drug induced QT prolongation and the likelihood of the development of TdP, which can occasionally occur without any substantial prolongation of the QT interval.
QTc values for normal and prolonged QT interval after correction with Bazett’s formula6
The QT interval is influenced by heart rate. The RR interval preceding the QT interval should be measured for rate correction. Several formulae may be used to correct the QT interval for the biophysical effect of heart rate (QTc), but none is perfect. The most commonly used formulae are Fridericia’s cube root formula (QTc = QT/RR1/3 ) and Bazett’s square root formula (QTc = QT/RR1/2). Of the two, Bazett’s formula is the more popular, but Fridericia’s correction is preferred because it is more accurate at the extremes of physiological heart rate.w4 w5 Apart from heart rate, the duration of the QT interval is also subject to the techniques of recording and measurement error of the QT interval, sympathovagal activity, drugs, genetic abnormalities, electrolyte disorders, cardiac or metabolic diseases, changes of cardiac afterload, and diurnal variation which can be up to 75–100 ms. It is important to remember that for every individual there is a different relation between the QT interval and the heart rate. Although the rate–correction formulae are useful clinically, they may not be accurate enough, especially when assessing the minor changes of the QT interval induced by drugs. The suggested QTc values using the Bazett’s formula for diagnosing QT prolongation are outlined in table 1.7
Newer repolarisation parameters such as QT dispersion (maximum – minimum QT intervals) on the 12 lead surface ECG, which is considered to be an indirect measure of spatial heterogeneity of repolarisation, may be useful in assessing drug efficacy and safety. In one important study, patients who received class 1a antiarrhythmic drugs and developed TdP had significantly increased precordial QT interval dispersion.w6 In contrast, patients receiving amiodarone or class 1a antiarrhythmics without TdP did not have increased QT dispersion, although the QT interval was noticeably prolonged.w6 Thus, spatial heterogeneity/dispersion of the ventricular repolarisation process may be required in addition to QT prolongation for the genesis of TdP. Although the use of QT dispersion in the assessment of drugs that prolong the QT interval needs further confirmation, it may provide information about the clinical significance of QT prolongation.
DRUGS THAT CAUSE QT PROLONGATION AND/OR TORSADES DE POINTES
The list of drugs that can prolong QT interval and/or cause TdP is extensive (table 3).
Drugs that can prolong QT interval and torsades de pointes (this list is not comprehensive)
Antiarrhythmics
The early landmark report by Selzer and Wray observed that quinidine use was associated with syncope and ventricular fibrillation or flutter.w7 In their report, the risk of TdP with quinidine was not necessarily a consequence of excessive doses of the drug. Others have confirmed that TdP with class Ia drugs can occur at low therapeutic or subtherapeutic concentrations.w8 Indeed most of the class Ia drugs, including quinidine, disopyramide, and procainamide, are similar in this regard.w9 On the other hand, antiarrhythmic agents such as sotalol are associated with a greater incidence of TdP as the dose increases.w10 One possible explanation for such discrepancy is that the blockade of sodium channels by class Ia drugs suppresses the QT prolonging effect at higher concentrations. Pure IKr potassium blocking antiarrhythmic drugs such as d,l,-sotalol prolong the QT interval and induces TdP at an incidence directly proportionate to their concentration until the potassium currents are completely blocked.w9 The mean effect on QTc prolongation by d,l,-sotalol varies from 10–40 ms at doses from 160–640 mg/day.
It is noteworthy that while class Ia drugs are strongly concordant in their production of TdP, concordance with class III antiarrhythmic agents is less clear. For example, while both sotalol and amiodarone have the same potent effects on QT prolongation, the incidence of TdP is very low with amiodarone compared with sotalol. A literature review revealed that the incidence of TdP with amiodarone was only 0.7% in 17 uncontrolled studies (2878 patients) between 1982 and 1993, and that no pro-arrhythmia was reported in seven controlled studies (1464 patients) between 1987 and 1992.8 Indeed, the evidence from a recent meta-analysis of amiodarone trials showed that amiodarone actually reduced the risk of arrhythmic death and resuscitated cardiac arrest in patients after myocardial infarction or with heart failure.w11 The risk of TdP with amiodarone mainly occurs in patients with other co-existing risk factors such as hypokalaemia or bradycardia. In contrast, d,l,-sotalol has a 0.3% incidence rate of TdP for a daily dose of 80 mg, which rises to 3.8% for a daily dose of > 680 mg.w9 The risk is greater in female patients, and patients with reduced creatinine clearance, congestive heart failure, or sustained ventricular tachycardia. In the USA, patients initiated or re-initiated on d,l,-sotalol are required to be admitted to hospital for a minimum of three days (on their maintenance dose) in a facility that can provide cardiac resuscitation, continuous electrocardiographic monitoring, and calculations of creatinine clearance (that must precede administration of the first dose of d,l,-sotalol), and in the presence of personnel trained in the management of serious ventricular arrhythmias in order to minimise the risk of TdP.w12 The risk of TdP can be reduced by adjustment of the d,l,-sotalol dose according to creatinine clearance and by monitoring the ECG for excessive increases in the QT interval. d,l,-Sotalol is contraindicated in patients with creatinine clearance < 40 ml/min or QTc interval > 450 ms.w12
Similar to d,l,-sotalol, dofetilide also exhibits a dose dependent effect on QTc prolongation and TdP. The mean QTc prolongation with dofetilide varies from 5–20 ms at doses of 125–500 μg twice daily, and the incidence of TdP ranges from 0–10.5% at doses < 250 μg to > 500 μg.w13 Other new class III intravenous antiarrhythmics, such as ibutilide, are equally toxic in inducing TdP. In the ibutilide repeat dose study, 8.3% of the patients developed TdP during or soon after the start of two, 10 minute infusions, separated by 10 minutes of ibutilide (1.0 and 0.5 mg or 1.0 and 1.0 mg).w14 Similarly, in patients with atrial fibrillation, intravenous almokalant induced TdP in 6% of the patients.w15
Oral azimilide at doses up to 200 mg/day prolongs the QTc interval by 4–42%.w16 Several cases of TdP have been reported in association with azimilide use, usually when bradycardia, pauses, or hypokalaemia are present, but the preliminary results from the ALIVE (azimilide post-infarction survival evaluation) study showed that azimilide prescribed at 100 mg has a low incidence of TdP even in high risk post-myocardial infarction patients with reduced heart rate variability.w17
Antihistamines
Since 1986, certain non-sedating antihistamines, the so called second generation antihistamines (mainly terfenadine and astemizole), have been reported to cause QT prolongation and, in some cases, TdP.9w18 These incidents have occurred when the recommended dose has been exceeded, at normal doses with concurrent use of drugs that inhibit hepatic cytochrome P450 enzymes (for example, imidazole, antifungals, and macrolide antibiotics), impaired liver function, or in patients with congenital long QT syndrome.9 Like class III antiarrhythmics, terfenadine and astemizole were found to prolong the monophasic action potential and QT interval, which led to the development of early after-depolarisation and TdP through inhibition of the IKr channel.w19 As almost all of the non-sedating antihistamines were metabolised via the hepatic cytochrome P450 CYP3A4 system, concomitant administration of drugs or food (grapefruit juice) that inhibit the hepatic cytochrome P450 or severely compromise liver function may result in the accumulation of the parent drug and cardiotoxicity.w19 Furthermore, co-administration of non-sedating antihistamines with other drugs that prolong the QT interval by the same or other mechanism (for example, antiarrhythmics, antipsychotics, tricyclic antidepressants) also increases their adverse effect on cardiac repolarisation.w19 Other drug related factors such as the physicochemical properties of the antihistamines (for example, diarylalkylamine moiety, quaternisation of diphenhydramine, lipophilicity of the side chain), their metabolic profile, and tissue distribution may also contribute to the cardiac response of antihistamines.
Newer non-sedating antihistamines (loratadine, cetirizine, acrivastine, mizolastine, ebastine, and fexofenadine) continue to be introduced into the market. The cardiac safety profile of these newer non-sedating antihistamines will require confirmation. Antihistamines with low or no potential to block the K+ rectification channel (for example, IKr) channels are likely to possess cardiac safety advantages. The overall evidence so far indicates that the potential to cause TdP is not a class effect of non-sedating antihistamines; certain non-sedating antihistamines such as terfenadine and astemizole have potent pro-arrhythmic risk, whereas others have low risk of (for example, azelastine, mizolastine) or are probably not associated with (for example, loratadine, cetirizine, ebastine and fexofenadine) QT prolongation, TdP or other ventricular arrhythmias. It should be emphasised that apart from the specific contraindications described, the incidence of cardiotoxicity with antihistamines is very low in view of the widespread use of the drugs.10 Nevertheless, as they are widely prescribed for a self limiting, non-fatal disease, the risk attributable must be assessed very carefully.
Antimicrobials
Macrolides (erythromycin, clarithromycin), fluoroquinolones, antifungals, and antimalarials have been implicated in predisposing to TdP as a result of QT prolongation.11w20–23 These reports are few and anecdotal. Similar to class III antiarrhythmics and antihistamines, macrolides prolong the QT interval and cause dispersion of repolarisation across the ventricular wall, resulting in the induction of TdP. In the case of fluoroquinolones, sparfloxacin lengthened the duration of the action potential in a concentration dependent manner,12 whereas ofloxacin and levofloxacin did not alter the action potential duration at a variety of concentrations (1–100 μM).w24 Thus, sparfloxacin exerts a pure class III electrophysiological effect whereas levofloxacin and ofloxacin do not. Clinically, it is not yet known whether sparfloxacin will cause any spontaneous TdP, particularly in low risk patients. Since the drug was marketed in 1994, there were very few cases of ventricular arrhythmia (three reversible ventricular tachycardias) reported during the European post-marketing surveillance of sparfloxacin, all of which occurred in patients with underlying cardiac conditions.12 Grepafloxacin, a new fluoroquinolone available in the UK since 1998, has recently been withdrawn voluntarily by its distributor because of its effect on QT prolongation and some reported cases of TdP. The available evidence from preclinical and clinical studies suggested that there are significant differences in the potency to prolong QT interval among the fluoroquinolones, and the risk of arrhythmias varies between drugs13(fig 4). Sporadic cases of TdP have been reported in association with most, but not all, fluoroquinolones. As a whole, apart from grepafloxacin and possibly sparfloxacin, the fluoroquinolones that are currently on the market or soon to be launched are safe from the point of view of QT prolongation and TdP, with a frequency of this adverse event generally occurring at a rate of about one per million prescriptions.

Effect of various fluoroquinolones on prolonging action potential duration. Modified and reproduced from Hagiwara and colleagues13 with permission.
Antimalarials deserve some attention as they are commonly prescribed worldwide. Quinine, quinidine, and halofantrine are capable of prolonging the QT interval.w25–28 Quinine prolongs the QT interval at standard doses, as does halofantrine (60%). Halofantrine induces a dose related prolongation of the QT interval whereas mefloquine has no effect on QT interval.w25 However, the risk of significant QT prolongation (> 25% or QTc ⩾ 0.55 s1/2) was greater if halofantrine was given as a re-treatment following mefloquine failure than as primary treatment.w25 The Committee of Safety of Medicines in the UK recommends that halofantrine should not be given with other drugs that prolong QT interval or to patients with any form of cardiac condition associated with QT prolongation. Cardiotoxicity of antimalarials is increased in patients with acute renal failure, especially after three days of treatment.w26 Hence, it has been recommended that ECG monitoring should be carried out during quinidine infusion.w26 However, it is interesting to note that pre-treatment ECGs were poorly predictive of QT prolongation during oral treatment of halofantrine, although it may be useful for evaluating patients with pre-existing cardiac conditions.w27 This is partly because the degree of QT prolongation is dependent on the plasma concentration of halofantrine.w28
The antifungal agents ketoconazole and itraconazole prolong the QT interval by blocking the IKr channels.w29 w30 Similar to macrolide antibiotics, ketoconazole and itraconazole also inhibit the hepatic cytochrome P450 CYP3A4 isoenzyme.w29 w30 Therefore, co-administration of ketoconazole or itraconazole with another QT prolonging drug that is metabolised by the cytochrome P450 CYP3A4 isoenzyme, such as terfenadine, will result in a notably prolonged QT interval and increase the risk of TdP.w29–31
Tricyclic antidepressants
The use of tricyclic antidepressants (TCAs) has raised some concern about their cardiotoxicity. The effect of TCAs on the QT interval have been investigated, but with mixed results.w32–34 Amitriptyline, doxepin, desipramine, imipramine, and clomipramine have been associated with a prolonged QT interval, whereas dothiepin has no effect on QT interval. In children, concerns about possible TCA associated adverse effects were raised after a few cases of sudden death in children treated with TCAs.w35 w36 The TCAs implicated were desipramine, clomipramine, and imipramine. These cases of sudden death occurred without acute overdose. A possible mechanism is the “fast” or “slow” metabolism of TCA by hepatic cytochromes.14w37 For example, impaired metabolism caused by a genetically determined “slow metaboliser” phenotype of cytochrome CYP2D6 is suggested as a possible mechanism for the apparent toxicity of these tricyclic antidepressants.w37 Co-administration of drugs that can alter the concentrations of both parent drug and metabolites will therefore affect the QTc interval. It has been recommended that children and adolescents on TCA have an ECG at baseline and after each dose increase.w38
Recently, there have been case reports of abnormal ECG changes, akin to that observed in patients with Brugada’s syndrome, following overdoses of antidepressants and neuroleptics (namely, phenothiazine, amitriptyline, fluoxetine), and therapeutic doses of trifluoperazine and loxapine,w39 w40 with tricyclic antidepressants being the most frequently implicated. The ECGs in these patients showed right bundle branch block and ST segment elevation on the precordial leads, similar to that seen in patients with Brugada’s syndrome, following the ingestion of these drugs. These changes, however, disappeared after the withdrawal of the drugs and could not be reproduced with the subsequent flecainide tests on these patients. Evidence from cellular studies suggest that, similar to class Ic drugs, amitriptyline, phenothiazine, and fluoxetine induce cardiac sodium channel blockade and reduce Ito activation, which may shorten the action potential durations and induce an intramyocardial electrical gradient that produces the typical ECG changes described above. However, such ECG changes will probably only occur upon massive overdose of these drugs as in the case of these patients, which may explain why the ECG changes could not be reproduced with subsequent flecainide challenge. Furthermore, it is also possible that these patients may have subclinical dysfunctional sodium channels that were unmasked by these drugs. Thus, it has been postulated that this could be another mechanism for drug induced sudden death in patients receiving chronic treatment with tricyclic antidepressants and neuroleptics. Nevertheless, further studies are required to investigate this phenomenon.
Neuroleptics
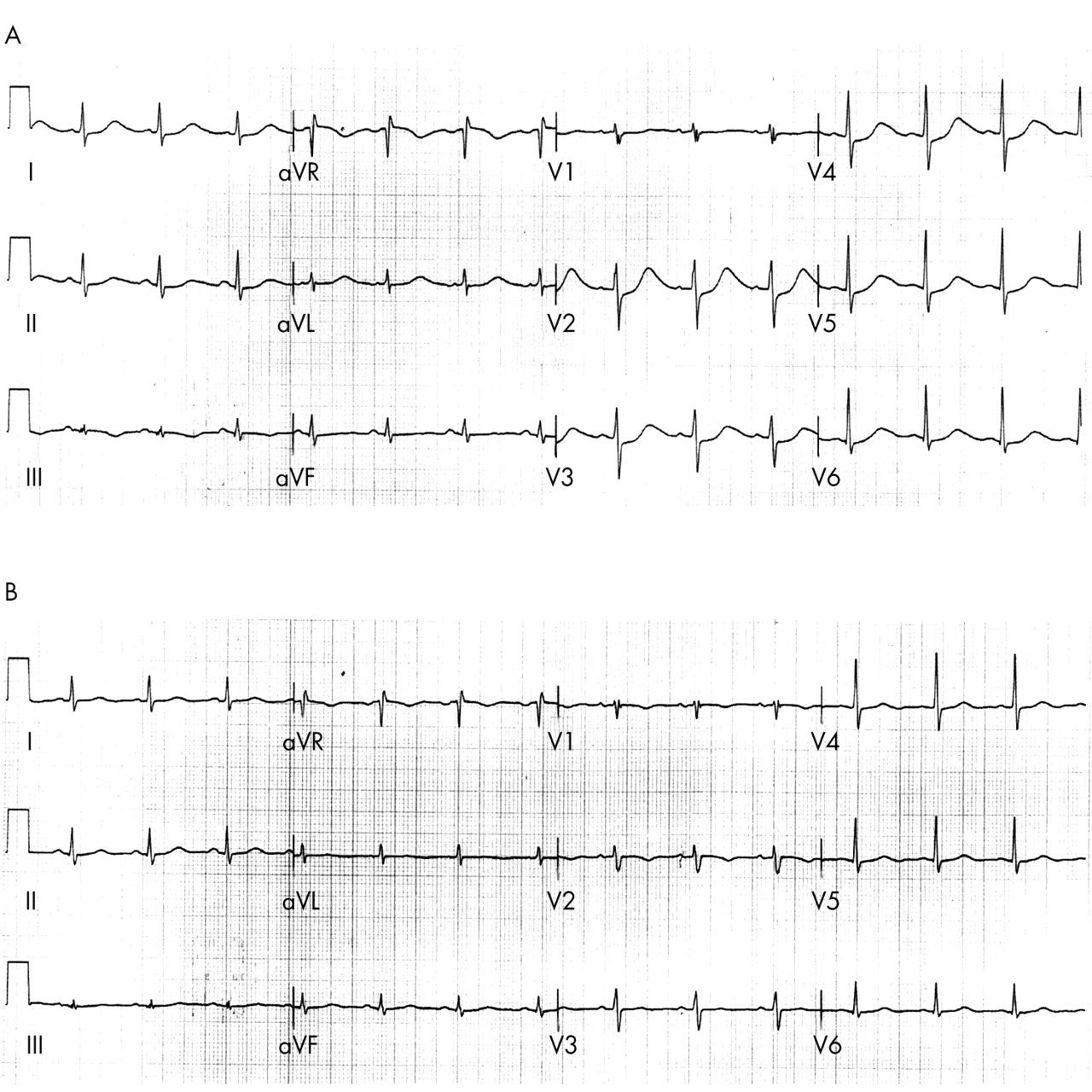
Neuroleptics have long been associated with sudden death and are reported to cause QT prolongation and TdP at therapeutic doses or in overdose (phenothiazines, thioridazine, haloperidol, chlorpromazine, trifluoperazine, pericycline, prochlorperazine, and fluphenazine).15w41–45 Among them, thioridazine was the most potent in causing QT prolongation and arrhythmia15 (fig 5). In addition, the clinical use of thioridazine has also been known to cause a decrease in T wave amplitude, and a prominent U wave in approximately 50% of patients receiving 100–400 mg/day of the drug, albeit seldom with clinical consequences. At toxic concentrations, thioridazine can cause sinus bradycardia, atrioventicular block, pronounced QT prolongation, and recurrent ventricular tachycardia and fibrillation.w46 At both therapeutic and toxic doses, thioridazine can induce TdP.w42 w46 In the presence of hypokalaemia, TdP can develop even with a low dose (50 mg daily) of thioridazine.w43
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
(A) The ECG of a middle aged woman who was otherwise healthy but suffered a ventricular fibrillation cardiac arrest while receiving 20 mg daily of thioridazine. This ECG was recorded immediately after the cardiac arrest. Note the prolonged T wave offset resulting in a prolonged QTc interval of 619 ms. (B) The ECG of the same patient three days after the withdrawal of thioridazine (QTc = 399 ms).
Sertindole is a relatively new atypical antipsychotic for the treatment of schizophrenia. Its safety and efficacy were assessed in three double blind randomised studies in the USA, North America, and Europe.w47 Slight QT prolongation was seen with sertindole in early clinical trials although TdP was not reported in these studies. However, 12 unexplained sudden deaths and 23 cases of syncope occurred among 1446 patients during the pre-marketing trials of sertindole.w48 A total of 27 deaths associated with its use had been reported to the US Food and Drug Administration (FDA) by 1996. Although an independent review panel then did not find a causal relationship between sertindole and these deaths,w49 in 1996 the drug was not approved for marketing in the USA.
Nevertheless, sertindole was marketed in Europe. However, in the UK the Committee of Safety of Medicines was notified of 36 suspected adverse drug reactions with a fatal outcome by the end of November 1998.w50 Not all of these reports were related to sudden cardiac death. In addition, 13 reports of serious but non-fatal cardiac arrhythmia were also reported in the UK during the same period. Because of the number of adverse drug events, fatal and non-fatal, reported since the marketing of sertindole in the UK, it was considered that the risks of treatment with this drug outweighed its benefits. The manufacturers of sertindole voluntarily suspended its marketing and use from December 1998 in the UK, pending further safety evaluations. It is now known that sertindole is a high affinity antagonist of the human cardiac IKr potassium channel, and this blockade underlies, at least in part, the prolongation of the QT interval observed with this drug.16
Pimozide is a diphenylpiperidine neuroleptic agent with known cardiovascular side effects including QT prolongation.w51 TdP has been described after acute poisoning.w52 The risk of pimozide cardiotoxicity may be increased with the concomitant use of drugs that inhibit the cytochrome P450 CYP3A4 isoenzyme–for example, clarithromycin, ketoconazole, etc.w53 Forty reports (16 fatal) of serious cardiac reactions (predominantly arrhythmias) with pimozide use were reported to Committee of Safety of Medicines between 1971 and 1995,17 and restricted labelling has now been introduced for pimozide in the UK.
Prokinetics
Cisapride is gastrointestinal prokinetic agent used to treat gastro-oesophageal reflux, gastroparesis, and childhood chronic intestinal pseudo-obstruction; it is structurally similar to procainamide. In the USA, while cisapride was being marketed from 1993 to 1999, the FDA received reports on a total of 341 individual patients who had serious adverse cardiac effects following the use of cisapride: 117 developed QT prolongation; 107 TdP; 16 polymorphic ventricular tachycardia; 27 ventricular tachycardia; 18 ventricular fibrillation; 25 cardiac arrest; 16 serious (unspecified) arrhythmia; and 15 sudden death.18 Eighty (23%) of the 341 patients died. Deaths were directly or indirectly associated with an arrhythmic event. Many of the patients (56%) were also taking an imidazole compound or a macrolide antibiotic, which could inhibit the P450 CYP3A4 isoenzyme that metabolises cisapride and results in increased serum concentrations.
In the UK, since cisapride was first marketed in 1988 the Medicines Control Agency has received reports of 60 serious cardiac adverse drug reactions, five of which were fatal. These included 24 reactions comprising ventricular arrhythmias, sudden unexplained death, cardiac arrest, and QT prolongation.w54 Worldwide, there have been 386 reports of serious ventricular arrhythmias associated with cisapride treatment, 125 of which were fatal, and 50 reports of sudden unexplained death. In the UK, several relabellings of cisapride were required following these incidences, which had a limited effect in reducing co-prescription of cisapride with contraindicated medication. Serious cardiovascular reactions, including fatalities, continued to be reported. As a result, the Medicines Control Agency suspended the product licences for cisapride in the UK in July 2000 as the risks versus benefits balance was no longer considered favourable. Similarly, in the USA, the risk of fatal arrhythmia with cisapride was believed to outweigh the benefit for the approved indication, treatment of nocturnal heartburn, leading to the drug’s discontinuation there. Cisapride inhibited the IKr current in isolated guinea pig ventricular myocytes in a concentration dependent manner with an IC50 of 15 nmol/l (therapeutic levels, 50–200 nmol/l).w55 This explained the lengthening of cardiac repolarisation observed in patients receiving clinical doses of cisapride.
Other QT prolonging drugs that have been withdrawn
Early reports of TdP associated with cardiac drugs incriminated not only antiarrhythmics, but antianginal agents such as bepridil and prenylamine, both of which have been well documented to cause TdP.w56 w57 These antianginal agents have now been withdrawn from the market in most regulatory jurisdictions. Terodiline, an antispasmodic agent used to treat urinary incontinence, was withdrawn in the UK following 69 reported cases of serious arrhythmias. Fourteen of these patients had sudden death and the remaining 55 patients had non-fatal arrhythmias, including 37 with ventricular tachyarrhythmia of which 24 were caused by TdP.w58 It is now clear that the pro-arrhythmic effect of terodiline is a consequence of the blockade of IKr current,w59 which occurs in a concentration dependent manner.
OTHER FACTORS THAT MAY INCREASE THE PROLONGATION OF VENTRICULAR REPOLARISATION OR PREDICT TORSADES DE POINTES
Apart from drugs, other conditions that are likely to cause QT prolongation include:
-
organic heart disease (for example, congenital long QT syndrome, ischaemic heart disease, congestive heart failure, dilated cardiomyopathy, hypertrophic cardiomyopathy, myocarditis, and Kawasaki syndrome)w60–64
-
metabolic abnormalities (for example, hypokalaemia (by far the most common), hypocalcaemia, hypomagnesaemia)w65–67
-
bradycardia, atrioventricular and sinoatrial blocksw68 w69
-
drug related factors (for example, narrow therapeutic window, a multiplicity of pharmacological actions and inhibition and induction of cytochrome P450 enzymes, polypharmacy)19w70
-
female preponderance, which may be caused by sex differences in specific cardiac ion densitiesw71 w72
-
hepatic impairment.
PREVENTION OF DRUG INDUCED QT PROLONGATION
In clinical practice, adverse effects of QT prolonging drugs can be prevented by not exceeding the recommended dose, avoiding their use in patients with pre-existing heart disease or risk factors as mentioned above, previous ventricular arrhythmias, and/or electrolyte imbalance such as hypokalaemia. Concomitant administration of drugs that inhibit the cytochrome P450 (for example, imidazole antifungals, macrolide antibiotics) or those that can prolong the QT interval or drugs that cause electrolyte disturbance should be avoided. The serum potassium concentration should be checked regularly as a matter of routine care when the patient is on potassium wasting diuretics. Furthermore, it may be sound clinical practice to perform ECGs routinely before and after an initiation or increment of dosage of a drug that may prolong the QT interval. If the patient develops TdP, the offending drug should be stopped and electrolyte abnormalities corrected. Drugs that can prolong the QT interval should ideally be listed and regularly updated in a national drug formulary, which is not the case at present. Any adverse event suggestive of cardiac arrhythmias should be reported urgently to drug safety authorities and/or drug manufacturers.
The management of patients with drug induced TdP includes identifying and withdrawing the offending drug(s), replenishing the potassium concentration to 4.5–5 mmol/l, and infusing intravenous magnesium (1–2 g). In resistant cases, temporary cardiac pacing may be needed to increase the heart rate and shorten the QT interval.
REGULATORY PERSPECTIVE IN DRUG DEVELOPMENT
Apart from antiarrhythmics, many drugs capable of inducing TdP are non-cardiac and are used for relatively benign conditions. Regulatory authorities in the European Union (EU) are now concerned that the risk should be identified and if possible quantified during the preclinical and clinical development of a drug. Currently there are no contemporary guidelines from other regulatory authorities to address this issue. In 1997, the UK Committee for Proprietary Medicinal Products (CPMP) adopted a document entitled Points to consider: the assessment of the potential for QT interval prolongation by non-cardiovascular medicinal products.20 The CPMP guideline document should be viewed as a strong signal from the public health authorities that the problem of QT prolongation, especially by non-cardiac drugs, is significant and requires careful scrutiny. Additional research and development are needed for any compound with the potential to prolong the QT interval. The CPMP document details the necessary preclinical and clinical stages required for testing the safety of new active substances.
CONCLUSION
It has been well recognised that a prolonged QT interval (congenital or acquired) on the surface ECG is associated with an increased risk of TdP and/or sudden death. By far the most common cause of acquired long QT syndrome is drug induced, with antiarrhythmics being the group of drugs most commonly implicated. Since the 1990s, seven non-cardiac drugs marketed in the UK—namely, terfenadine, astemizole, cisapride, terodiline, halofantrine, sertindole, and pimozide—have attracted regulatory attention because of their propensity to produce QT prolongation, TdP, and/or sudden death. The list of drugs, especially non-cardiac drugs, which can cause some degree of QT prolongation may continue to grow. The risk of TdP is therefore likely to remain a significant problem in the future. All physicians and pharmacists, and patients who receive these drugs, should be made aware of this risk and educated accordingly, and take precautions to minimise pro-arrhythmia. Preclinical and clinical evaluations remain the cornerstone for assessing the arrhythmogenic potential of any new drug before approval. Finally, post-marketing surveillance is also important for monitoring spontaneous adverse cardiac effects.
Drug induced QT prolongation and torsades de pointes: key points
-
Drug induced QT prolongation and torsades de pointes are an increasing public health problem
-
The blockade of IKr potassium current by these drugs is responsible for their pro-arrhythmic effect
-
Measurement of QT interval should be corrected for heart rate
-
Antiarrhythmic drugs, non-sedating antihistamines, macrolides antibiotics, antifungals, antimalarials, tricyclic antidepressants, neuroleptics, and prokinetics have all been implicated in causing QT prolongation and/or torsades de pointes
-
Co-administration of multiple drugs, especially with other QT prolonging drug(s) and/or hepatic cytochrome P450 CYP3A4 isoenzyme inhibitors, must be avoided
-
The risk of QT prolongation is increased in females, patients with organic heart disease (for example, congenital long QT syndrome, myocardial infarction, congestive heart failure, dilated cardiomyopathy, hypertrophic cardiomyopathy, bradycardia), hypokalaemia, and hepatic impairment
-
The treatment of drug induced torsades de pointes includes identifying and withdrawing the offending drug(s), replenishing the potassium concentration to 4.5–5 mmol/l, and infusing intravenous magnesium (1–2 g). In resistant cases, temporary cardiac pacing may be needed
REFERENCES
Supplementary materials
. Web-only References
These references are available as a downloadable PDF (printer friendly file).If you do not have Adobe Reader installed on your computer,
you can download this free-of-charge, please Click hereFiles in this Data Supplement:
Linked Articles
- Miscellanea
- Miscellanea
- Miscellanea
- Miscellanea
- Miscellanea
- Miscellanea
- Miscellanea
- Miscellanea
- Miscellanea
- Miscellanea