Article Text

Abstract
Introduction Recent studies showed the usefulness of globotriaosylsphingosine (lyso-Gb3) and related analogues, deacylated forms of globotriaosylceramide (Gb3), for high-risk screening, treatment monitoring and follow-up for patients with Fabry disease.
Methods We evaluated Gb3, lyso-Gb3 and analogues using tandem mass spectrometry in 57 women with Fabry disease followed during a period of 15.4 years. Twenty-one women were never treated and 36 received treatment (agalsidase-beta, n=30; agalsidase-alfa, n=5; or migalastat, n=1). Lyso-Gb3 and analogues at m/z (−28), (−2), (+16), (+34) and (+50) were analysed in plasma and urine. Total Gb3 and lyso-Gb3 analogues at m/z (−12) and (+14) were evaluated in urine while the analogue at m/z (+18) was evaluated in plasma.
Results A strong correlation between plasma and urine lyso-Gb3 and analogue levels was revealed. Plasma and urine lyso-Gb3 and analogue levels were not statistically different between patients carrying missense (n=49), nonsense (n=6) or deletion mutations (n=2). Never treated patients had lower plasma lyso-Gb3 and analogues at m/z (−28), (−2), (+16), (+34) and the seven urinary lyso-Gb3 analogues compared with pretreatment levels of the treated patients. A significant reduction of plasma lyso-Gb3 and five analogues, as well as urine Gb3 and six lyso-Gb3 analogues, but not lyso-Gb3 and lyso-Gb3 at m/z (+50), was observed post-treatment with agalsidase-beta. The same tendency was observed with agalsidase-alfa.
Conclusion Women with Fabry disease who started treatment based on clinical manifestations had higher lyso-Gb3 and analogue biomarker levels than never treated women. This indicates that a biomarker cut-off could potentially be a decision tool for treatment initiation in women with Fabry disease.
- genetic research
- genetics
- medical
- genotype
- human genetics
- phenotype
Data availability statement
All data relevant to the study are included in the article or uploaded as supplementary information. This research project involves prospective data (15 years) from a retrospective cohort. Patients were part of the nationwide Danish long-term clinical Fabry Disease cohort.
Statistics from Altmetric.com
Introduction
Fabry disease (FD, OMIM no. 301500) is an X linked inherited lysosomal storage disorder caused by mutations in the GLA gene encoding for the alpha-galactosidase A enzyme (α-GAL; EC 3.2.1.22).1 This protein is involved in the catabolism of large molecules, such as glycosphingolipids. FD is characterised by the accumulation of several glycosphingolipids, such as globotriaosylceramide (Gb3),2 3 galabiosylceramide (Ga2),4 and the deacylated form of Gb3, globotriaosylsphingosine (lyso-Gb3)5 6 in the vascular endothelium, tissues of various organs and biological fluids. Progressive intracellular accumulation leads to tissue damages and eventually to organ dysfunction, reduced quality of life and premature death.
Fabry manifestations are multisystemic and progressive: acroparesthesia, diaphoresis, gastrointestinal symptoms and proteinuria are frequent in adolescents and even in children, while adults are prone to major organ involvement including impaired renal function, cardiomyopathy and cerebrovascular events.1 Despite Fabry being an X linked disorder, heterozygous women are carriers of the disease and may exhibit significant organ involvement, although usually at a later onset than men.7–10 Since 2001, enzyme replacement therapy (ERT) is available for treatment of FD. Two ERT medications are commercially available and administered by infusion: agalsidase-alfa (Replagal, Shire/Takeda Pharmaceutical) at 0.2 mg/kg body weight and agalsidase-beta (Fabrazyme, Sanofi Genzyme) at either 1.0 or 0.3 mg/kg body weight every other week.11 Studies support a benefit of ERT in women since ERT was associated with reduced albuminuria and glomerular/tubular urinary protein markers in women with FD with mild renal manifestations.12 Recently, a systematic literature review led by experts on FD concluded that ERT is associated with significant reduction in Gb3 accumulation and improvement of cardiac variables and quality of life in women.13 Oral pharmacological chaperone therapy with migalastat (Galafold) is a new treatment available for patients with an amenable mutation as an alternative to ERT.14
The phenotypic diagnosis of FD requires a combination of clinical, biochemical and molecular criteria, as well as biomarkers with the best available sensitivity and specificity to classify individual patients with respect to severity of disease, prognosis and management of treatment. Determination of Gb3, the main substrate of the α-GAL, in plasma or urine, has been used for a long time as a diagnostic tool in men following an abnormal α-GAL activity result and in women with inconclusive α-GAL activity measurements. However, Gb3 determination presented some limitations: (1) plasma or urine Gb3 concentrations were not always increased in heterozygotes and hemizygotes with non-classical mutations;15 (2) an overlap in plasma Gb3 levels was observed between the non-classic phenotype men, heterozygote women and healthy controls;16 (3) Gb3 has been found elevated in plasma and/or urine in pathologies other than FD.17 18
More than 10 years ago, lyso-Gb3, the more polar deacylated metabolite of Gb3, has been introduced as a biomarker of FD. Higher elevation of lyso-Gb3 concentrations and a better correlation with the phenotype in comparison with Gb3 was observed.16 Nevertheless, normal lyso-Gb3 concentrations cannot exclude the diagnosis in non-classic male patients, considering that lyso-Gb3 concentrations in patients with FD could overlap with the controls.19 Therefore, more sensitive and reliable biomarkers are strongly needed for screening and diagnosis of patients with FD on the one hand and monitoring of treatment management on the other. Recently, development in the field of metabolomics led to the discovery and chemical structure identification of lyso-Gb3 analogues in plasma and urine using a time-of-flight mass spectrometry approach.20 21 As confirmed by tandem mass spectrometry and exact mass measurements, lyso-Gb3 analogues have modifications on the sphingosine moiety of the molecule. Some of these analogues have shown interesting associations with clinical manifestations of FD, such as left ventricular mass index and Mainz Severity Score Index in patients with a late-onset IVS4 +919G>A cardiac variant mutation in Taiwan.22 Another study in a child cohort and in patients with the c.644A>G (p.N215S) cardiac variant mutation emphasised that lyso-Gb3 analogues were more sensitive biomarkers than lyso-Gb3 itself, especially in women.23 Although the α-GAL activity/lyso-Gb3 ratio in dried blood spots has been described as a more sensitive biomarker in diagnosing women with FD24 and an increase in plasma lyso-Gb3 seemed to be a marker of disease burden in treatment-naïve female patients with FD,25 the marked heterogeneity of the phenotype and genotype in FD shows that clinical and translational research studies are needed to better understand the biochemistry and pathophysiology involved, particularly in FD women.
Considering these unmet needs, we have devised a study to investigate a large cohort of 57 female patients with FD who were part of the nationwide Danish long-term clinical FD cohort focusing on a longitudinal biomarker evaluation aiming at three main objectives: (1) to evaluate total Gb3 and lyso-Gb3 and its analogues in urine and plasma of never treated and treated Danish female patients with FD over a period of 15 years; (2) to perform correlation studies between these biomarkers and the genotype of the female patients and (3) to establish correlations between these biomarkers and long-term ERT or chaperone therapy treatments.
Materials and methods
Patients
Fifty-seven heterozygous women of various ages with genetically confirmed diagnosis of FD were included in this audit of prospective data from a retrospective cohort. Patients were part of the nationwide Danish long-term clinical FD cohort. Data from this cohort have previously been published in part.12 26–36 Patients were either never treated (n=21, available plasma/urine specimens 20/19) or treated (n=36, available plasma/urine specimens pretreatment 33/14, post-treatment 35/34). Figure 1 shows a cohort diagram of treated and never treated female patients with FD under study. Decision for treatment initiation was made according to the recommendations described in the consensus by the European Fabry Working Group.37 Thirty treated patients received agalsidase-beta as first treatment (available plasma/urine samples pretreatment 27/11, post-treatment 29/24), five patients agalsidase-alfa (available plasma/urine samples pretreatment 5/2, post-treatment 5/4) and one patient migalastat (available plasma/urine samples pretreatment 1/1, post-treatment 1/1). Twelve patients continued with the same medication during the entire study period, while the rest switched to another treatment or stopped treatment (online supplemental figure 1).
Supplemental material

Cohort diagram of treated and never treated female patients with Fabry disease under study.
FD was confirmed in all patients by direct sequencing of all seven exons of the GLA gene. Forty-nine patients were carriers of a missense GLA variant (available plasma/urine specimens: 47/28), six of a nonsense variant (available plasma/urine specimens: 5/3) and in two women a deletion mutation was present (available plasma/urine specimens: 1/2). The following variants were observed: p.Gly85Asp (G85D—15 patients), p.Arg112Cys (R112C—10 patients), p.Ala156Thr (A156T—9 patients), p.Ile232Thr (I232T—6 patients), p.Asn34Ser (N34S—4 patients), p.Arg342Ter (R342X—3 patients), p.Gly271Ser (G271S—3 patients), p.Arg227Ter (R227X—2 patients), p.Tyr329SerfsTer19 (c.986delA—1 patient), p.Gly171Ser (G171S—1 patient), p.Arg301Ter (R301X—1 patient), p.Asn355Lys (N355K—1 patient), c.369+3_c.547+954del4096 (1 patient) (online supplemental table 1). Plasma and/or urine samples were collected during an overall period of 15.4 years (June 2001–November 2016). At least one plasma sample was available from 56/57 patients and at least one urine sample was available from 54/57 patients. The maximum available samples from a patient were 12 (online supplemental table 2). Matching urine and plasma samples collected at the same time were not always available.
Supplemental material
Supplemental material
Study design
The levels of Gb3, lyso-Gb3 and its various analogues among never treated patients (NTr group) and patients who received treatment (Tr group) at two different timepoints, prior to (BTr) and after treatment (ATr) were compared. Pretreatment and post-treatment levels were compared overall (all available measurements at both points) or pairwise (available data from the same individual at both timepoints). In addition, in the Tr group, levels of Gb3, lyso-Gb3 and its various analogues before and after the initial treatment were compared among the various medications (overall and pairwise analyses) and at treatment switching.
Analysis of Fabry biomarkers
Specimen collection
Plasma samples were collected from patients with FD in lavender K2-EDTA vacutainer tubes and stored at −80°C until analysis. Urine samples were collected in universal plastic tubes and stored at −80°C until further processing without any centrifugation or filtration.
Urinary globotriaosylceramide (Gb3)
Eight Gb3 isoforms (C16:0, C18:0, C20:0, C22:1, C22:0, C24:1, C24:0 and C24:OH) were measured in dried urine spots (DUS) along with creatinine as part of a multiplex liquid chromatography tandem mass spectrometry (LC-MS/MS) method, as previously described.38 39 Results were expressed as the total ion count of Gb3 isoforms normalised to creatinine. Briefly, 1 mL of urine was deposited on a 5 cm diameter disk of Whatman-GE 903 filter paper with the internal standards and dried for at least 4 hours. DUS extraction was performed with 4 mL of methanol using an orbital shaker for 1 hour, followed by analysis of the extract by LC-MS/MS. The method showed good intra-assay and interassay precision with RSDs≤13.7%. The intra-assay and interassay accuracy was acceptable for total Gb3 with biases≤19.2%. The limit of quantification of total Gb3 is 0.05 µg/mL and the normal reference value is ≤25 µg/mmol creatinine.
Plasma and urine globotriaosylsphingosine (lyso-Gb3) and related analogues
The levels of the various lyso-Gb3 analogues in urine and plasma were determined by UPLC-MS/MS following a solid phase extraction purification procedure according to already published methods.40 41 Lyso-Gb3 and the analogues at m/z (−28), (−2), (+16), (+34) and (+50) were determined in both urine and plasma, while the lyso-Gb3 analogues at m/z (−12) and (+14) were determined only in urine and the lyso-Gb3 analogue at m/z (+18) only in plasma.
Statistical analysis
Data are reported as mean (SD) or median (first Q1 and third Q3 quartiles) depending on the distribution. Data distribution was determined by Kolmogorov-Smirnov, Shapiro-Wilk and Anderson-Darling tests. In order to achieve normality of data, different types of transformations were tested without always achieving normality. Various groups were compared using Mann-Whitney U test or the Kruskal-Wallis test when more than two groups were involved and paired data were compared using the non-parametric Wilcoxon’s Signed-Ranks Test. Statistical analyses were performed using SPSS V.25 and Minitab V.19 with statistical significance set at p<0.05.
Results
The mean age of the patients at the entrance of the study was 38.7 (SD 18.5) years (never treated patients: 42.5 (SD 19.7); treated patients: 36.6 (SD 17.7)). The mean age at onset of treatment was 37.1 (SD 17.5).
Correlations between plasma and urine biomarker levels
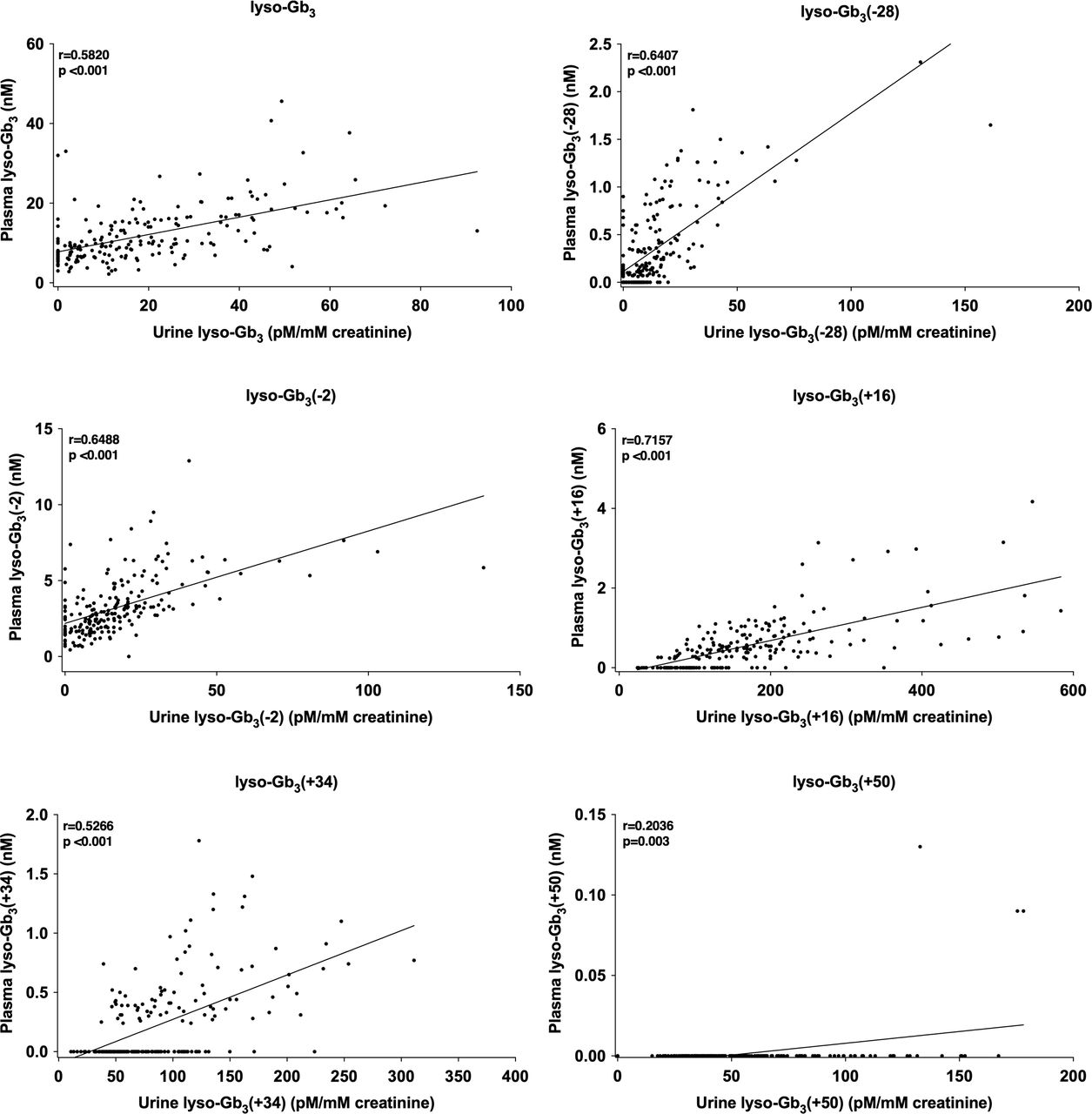
The correlations between urine and plasma lyso-Gb3 and its analogues (at m/z (−28), (−2), (+16), (+34) and (+50)) were evaluated when urine and plasma samples were collected at the same time or ±14 days. The analysis revealed a strong correlation between plasma and urine levels with all p<0.001 (figure 2).

Correlations between lyso-Gb3 and analogues in plasma and urine of female patients with Fabry disease (207 pair-samples). Pearson correlation: correlation is significant at the 0.01 level (2-tailed), r=Pearson correlation coefficient.
Association between GLA gene mutation classes and total Gb3, and lyso-Gb3 and analogues
No statistically significant different levels of plasma and urine lyso-Gb3 and its analogues as well as total urinary Gb3 were observed between the three GLA mutation classes (missense mutations, nonsense mutations, deletions). The same was true when the comparison was performed separately between never treated patients and the pretreatment levels of the treated patients with the exception of urinary lyso-Gb3 (+16). However, no reliable conclusions could be drawn due to the small sample size of the deletion mutations (one available plasma and urine specimens) and of the nonsense variants (available plasma/urine specimens: never treated patients 3/2, treated patients 2/1) (figure 3, online supplemental table 3).
Supplemental material

Descriptive comparisons of lyso-Gb3 and analogue levels in plasma and urine and Gb3 levels in urine, between GLA gene mutation classes (missense, nonsense, deletion) in never treated and treated patients (levels before treatment). The number above the end of the whisker represents the median, the end of the whisker represents the 75th percentile. Lyso-Gb3 (+50) data in plasma could not be analysed due to many results below LOD. LOD, limit of detection.
Plasma lyso-Gb3 and analogues
Never treated patients showed significantly lower levels of plasma lyso-Gb3 (median (Q1-Q3): 8.2 nM (6.1–11.0)) compared with pretreatment levels of treated patients (12.4 nM (9.4–20.0)) (figure 4, online supplemental table 4). The time interval between the date of biomarker measurement and the date of treatment is available in online supplemental table 4. The same was true for four lyso-Gb3 analogues (m/z (−28), (−2), (+16), (+34)), but not for (+18). Both overall and pairwise analysis revealed a significant reduction of plasma lyso-Gb3 and analogues post-treatment, in lyso-Gb3 at m/z (+18) and (+34) to below the limit of detection (LOD). Post-treatment levels were comparable with those from never treated patients. Lyso-Gb3 at m/z (+50) analogue data could not be analysed due to numerous values below LOD.
Supplemental material

Comparisons of lyso-Gb3 and analogue levels in plasma and urine, and Gb3 in urine between never treated and treated patients (overall analysis) and before and after treatment (pairwise analysis). The number above the end of the whisker represents the median, the end of the whisker represents the 75th percentile. Lyso-Gb3 (+50) data in plasma could not be analysed due to many results below LOD. LOD, limit of detection.
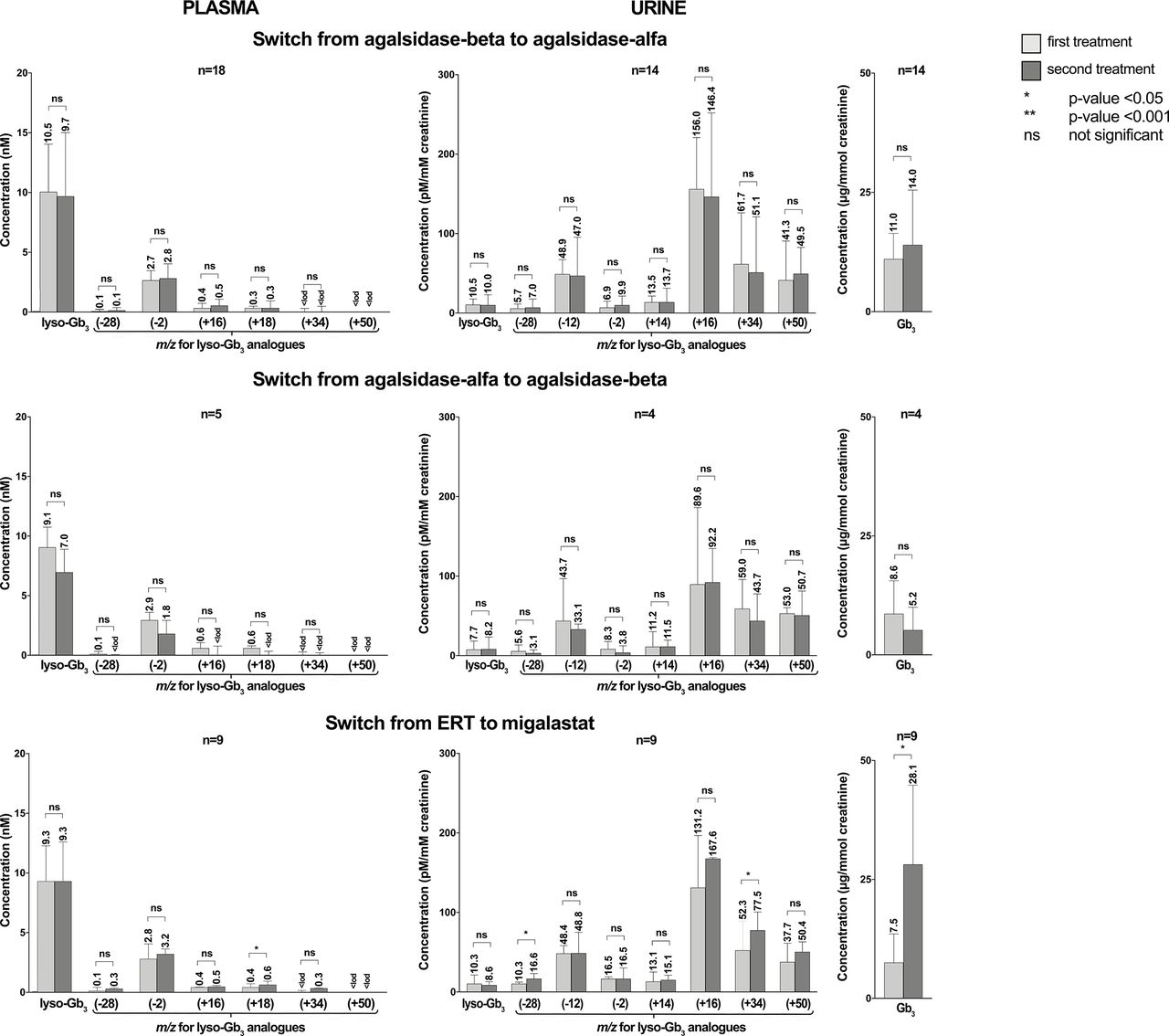
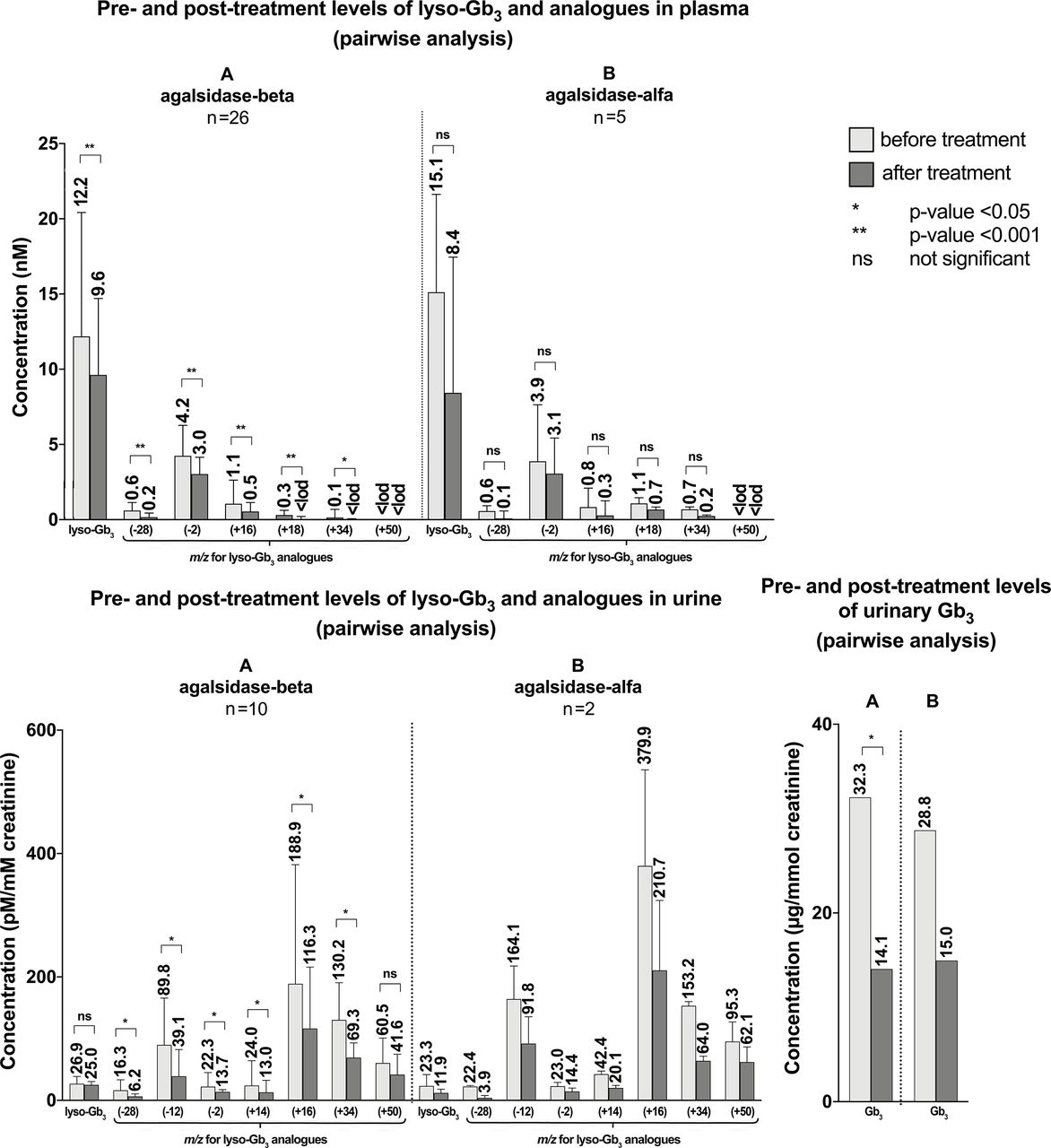
The pairwise analysis demonstrated a statistically significant reduction of lyso-Gb3 and five analogue levels ATr with agalsidase-beta (figure 5, online supplemental table 5). Statistical significance was not reached with agalsidase-alfa. However, the number of patients treated with agalsidase-alfa was smaller compared with those treated with agalsidase-beta (5 vs 26). Lyso-Gb3 at m/z (+50) data could not be analysed due to the numerous values below LOD. Reliable conclusions could not be reached for migalastat treatment due to the small sample size (one patient). No statistically significant differences were observed between lyso-Gb3 and analogues at treatment switching from agalsidase-beta to agalsidase-alfa and from agalsidase-alfa to agalsidase-beta. No differences were observed for lyso-Gb3 and four analogue levels from any ERT to migalastat, but this was not true for lyso-Gb3 analogue at m/z (+18). Nevertheless, numbers were too small for statistical performance, so the result is only descriptive (figure 6, online supplemental table 6).
Supplemental material
Supplemental material
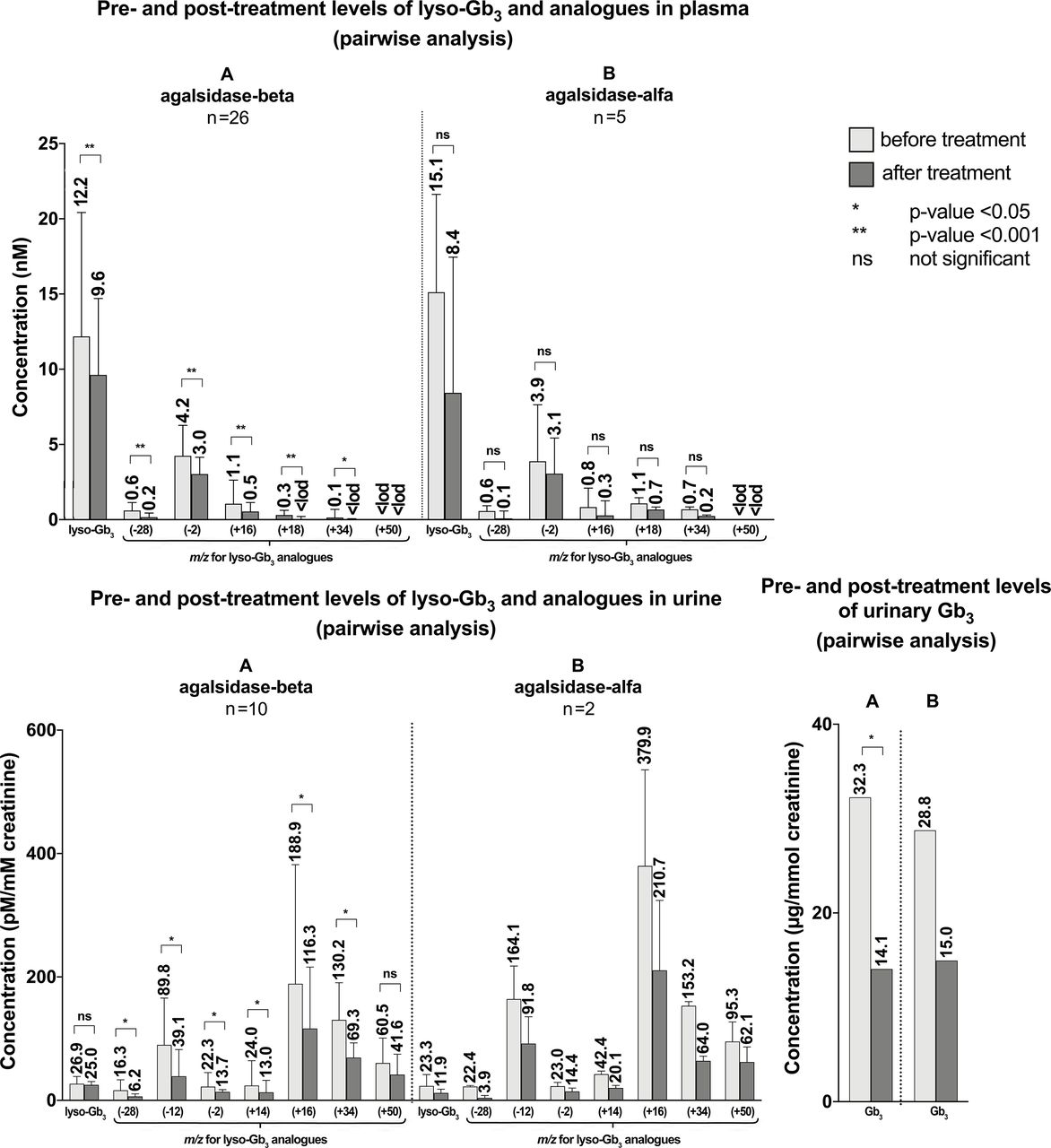
Pretreatment and post-treatment levels of lyso-Gb3 and analogues in plasma and pretreatment and post-treatment levels of Gb3, lyso-Gb3 and analogues in urine in patients who received treatment for Fabry disease. Pairwise analyses were used. The number above the end of the whisker represents the median, the end of the whisker represents the 75th percentile. (A) Treated with agalsidase-beta. (B) Treated with agalsidase-alfa. Lyso-Gb3 (+50) data in plasma could not be analysed due to many results below LOD. LOD, limit of detection.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Differences in the levels of lyso-Gb3 and analogues in plasma and urine, and Gb3 in urine at treatment-switching (pairwise analysis). The number above the end of the whisker represents the median, and the end of the whisker represents the 75th percentile. Lyso-Gb3 (+50) data in plasma could not be analysed due to many results below LOD. ERT, enzyme replacement therapy; LOD, limit of detection.
Urinary Gb3 and lyso-Gb3 and analogues
Never treated patients showed significantly lower levels of the seven urinary lyso-Gb3 analogues compared with pretreatment levels in treated patients (p<0.05) (figure 4, online supplemental table 4), but not of the urinary Gb3 and lyso-Gb3. Pairwise comparisons between pretreatment and post-treatment levels revealed a significant reduction of Gb3 and six lyso-Gb3 analogues at m/z (−28), (−12), (−2), (+14), (+16) and (+34). However, the concentrations of lyso-Gb3 and the analogue at m/z (+50) were comparable BTr and ATr (figure 4, online supplemental table 4).
The pairwise analysis showed a statistically significant reduction in post-treatment levels of urinary Gb3 and six lyso-Gb3 analogues after agalsidase-beta but not in lyso-Gb3 and the analogue at m/z (+50) (figure 5, online supplemental table 5). However, this analysis could not be performed for treatment with agalsidase-alfa (n=2) and migalastat (n=1) due to the small sample size. No statistically significant differences were observed between urinary Gb3, lyso-Gb3 and analogues neither at switching from agalsidase-beta to agalsidase-alfa nor from agalsidase-alfa to agalsidase-beta. Switching from any ERT to migalastat revealed an increase in the urinary Gb3 and the analogues of lyso-Gb3 at m/z (−28) and (+34) levels, but numbers were too small for statistical performance, so the result is only descriptive (figure 6, online supplemental table 6).
Discussion and conclusion
In the present study, we evaluated the profiles of lyso-Gb3 and related analogues and Gb3 of 57 women with genetically confirmed diagnosis of FD. We compared the lyso-Gb3 and analogue levels among never treated and treated patients pretreatment and post-treatment.
Our results showed that never treated patients have significantly lower levels of plasma lyso-Gb3 and the analogues at m/z (−28), (−2), (+16) and (+34) compared with the pretreatment levels of the patients who subsequently received treatment. The same was observed for urinary lyso-Gb3 analogues, but not for total urinary Gb3 and lyso-Gb3. We thus showed that women who eventually started treatment based on clinical manifestations had higher biomarker levels than never treated women. This indicates that a biomarker cut-off level might potentially be established as a decision tool for treatment initiation. Although studies demonstrated a decline in post-treatment plasma or urine Gb3 concentrations in patients with FD who received ERT and had elevated pretreatment Gb3 levels,42 43 posthoc analysis of prospective randomised clinical trials of agalsidase-alfa ERT in FD failed to detect a relationship between the reduction of Gb3 and prediction of clinical progression in patients with FD.44 In agreement with previous studies, we observed a significant reduction of plasma lyso-Gb3 levels after ERT.45 46 It is still unknown if this reduction correlates with the clinical outcomes ATr. On the contrary, the post-treatment urinary lyso-Gb3 levels were comparable with the pretreatment levels. In addition to plasma lyso-Gb3 reduction, we observed a significant reduction of plasma lyso-Gb3 analogues, six urine lyso-Gb3 analogues and urine Gb3 post-treatment, which reached comparable levels with those of the never treated patients.
We also compared the lyso-Gb3 and analogue levels BTr and ATr among the different types of treatment. Our results revealed significant reduction in post-treatment levels of plasma lyso-Gb3 levels after agalsidase-beta but not agalsidase-alfa treatment. We observed significant reduction in median post-treatment levels of five plasma lyso-Gb3 analogues after agalsidase-beta (lyso-Gb3 analogues at m/z (−28), (−2), (+16), (+18) and (+34), but not after agalsidase-alfa. This discrepancy between agalsidase-beta and agalsidase-alfa treatment could very likely be due to the small number of patients receiving agalsidase-alfa (five patients). A Dutch case control study revealed a significant decline 3 months after ERT, irrespective of the ERT preparation.45
No significant differences were observed in our study between plasma and urine lyso-Gb3 when ERT regimens were switched from one to the other, which is in keeping with three studies switching from agalsidase-beta to agalsidase-alfa during the agalsidase-beta shortage period.47–49 However, Smid et al 50 reported an increase in lyso-Gb3 in male patients after switching to agalsidase-alfa. No significant differences were observed in our study between plasma and urine lyso-Gb3 analogues after the switch. However, plasma lyso-Gb3 (+18) levels presented a tendency to increase in patients switching from agalsidase-beta to agalsidase-alfa while the opposite was the case in patients switching from agalsidase-alfa to agalsidase-beta (p=0.063 and 0.065, respectively), but the numbers on agalsidase-alfa were considerably smaller than the ones on agalsidase-beta. Taken together, these results and the observation that patients who were treated with agalsidase-beta but not those treated with agalsidase-alfa presented a significant reduction in plasma lyso-Gb3 at m/z (+18) levels, we can hypothesise that this latter analogue of lyso-Gb3 might be a reliable biomarker for treatment monitoring of patients. However, more studies are needed to investigate this hypothesis. An overview of changes in lyso-Gb3 and its analogue concentrations in plasma and urine in six female patients of different ages from the cohort are displayed in online supplemental figures 2 and 3. Interestingly, we can observe that the levels of plasma lyso-Gb3 and the analogue at m/z (−2) were still increased in most of the patients ATr independently of their age, whereas the rest of the analogues showed reduced levels in most cases (online supplemental figure 2). Unfortunately, we did not have baseline values in urine for five of these six women. Nevertheless, the 40-year-old woman with baseline values showed reduced urinary levels of lyso-Gb3 and all related analogues and Gb3 after 87 months of ERT treatment and chaperone therapy (online supplemental figure 3).
Supplemental material
Supplemental material
The strengths of this study were the long follow-up period of a nationwide cohort of female patients with FD including the availability of pretreatment and follow-up post-treatment samples. A longitudinal biomarker evaluation including female patients with FD was possible, since the cohort consisted of female patients with FD identified by family screening and therefore comprised a cohort unselected by both genotype and phenotype. Mutations were mostly classic with missense and nonsense mutations and deletions in a few cases. Finally, the biochemical measurements of total Gb3 and lyso-Gb3 with related analogues were done with the same apparatus, by the same validated methods providing uniform processing and analysis of samples over a short period of time.
The weaknesses of the study are that we cannot exclude a treatment-related selection bias by patient choice; some of the treatments were poorly represented with small numbers (mainly agalsidase-alfa (n=5) and migalastat (n=1) versus agalsidase-beta (n=26)). However, the demonstration of similar levels of the biomarkers in the treated patients and the never treated patients indicated that the combined choice of treatment by the treating physician and the patients was the correct one.
In conclusions, an overall profile analysis of lyso-Gb3 and analogue biomarkers might be a promising tool for long-term monitoring of patients with FD since we found that the levels of these latter biomarkers in urine and plasma were significantly reduced ATr. The plasma analogue of lyso-Gb3 at m/z (+18) seemed to be more reliable when ERT treatment was switched in these female patients. However, in another study, the association between lyso-Gb3 at m/z (+18) and clinical manifestations of the disease was not observed in patients with cardiac variant mutations,22 and other urine lyso-Gb3 analogues such as (−28), (−12), (−2), (+14), (+16) and (+34) were significantly different BTr and ATr in the women with classic mutations. This corroborates with the cardiac variant mutations in the Taipei study.22
Thus, the previously mentioned short-term prospective study involved establishing correlations between total Gb3 and lyso-Gb3+analogues in urine and plasma specimens of female patients with FD and the clinical manifestations and severity of the disease. Long-term prospective studies remain to evaluate the same biomarker concentrations in women, men and children with FD in order to better understand the general pathophysiology of the disease.
Data availability statement
All data relevant to the study are included in the article or uploaded as supplementary information. This research project involves prospective data (15 years) from a retrospective cohort. Patients were part of the nationwide Danish long-term clinical Fabry Disease cohort.
Ethics statements
Ethics approval
This project was approved by the Research Ethics Board at the Capital Region, Rigshospitalet, Copenhagen University Hospital (Journal number: H-18024706), at the Danish Data Protection Agency (2014-641-0055) and at the Faculty of Medicine and Health Sciences at the Centre intégré universitaire de santé et de services sociaux de l’Estrie-Centre hospitalier universitaire de Sherbrooke (CIUSSSE-CHUS).
Acknowledgments
We would like to thank Waters Corporation for their continued technical and scientific support. The expert assistance of Research Biotechnician Casper Kok and Research Nurse Ira Hagen Petersen is gratefully acknowledged. Thanks are due to all colleagues of the Fabry team in the Danish Capital Region for providing expert results and follow-up of the patients and to all Danish Fabry patients and their families for their continuous participation.
References
Footnotes
Contributors GE: performed the writing of the manuscript and the analysis of data. UF-R: was key initiator and advisor of the project, responsible for compiling the patient samples for analysis and patient data, continuously evaluated the data and progress and revised the manuscript critically. ÅKR: was an advisor in the project, involved in the interpretation of data and revised the manuscript critically. PL: wrote the first draft of the introduction, evaluated and interpreted data and revised the manuscript critically. MA: performed mass spectrometry biomarker analyses. MB: performed mass spectrometry biomarker analyses and revised the manuscript. CA-B: participated in the design of the study, provided the mass spectrometry infrastructure for all biomarker analyses, performed the evaluation and interpretation of data, provided insight in the writing of the draft and revised the final version of the manuscript critically. All authors approved the final manuscript.
Funding This Investigator-Initiated Research was supported by a grant (IIR-DNK-001030) from Shire International GmbH, now part of Takeda Pharmaceutical. The research salary of UF-R was supported by the Novo Nordisk Foundation.
Disclaimer The funding organisation played no role in the design of the study, choice of enrolled patients, interpretation of data, nor in preparation of the manuscript.
Competing interests GE has received conference registration fees and travel grants from Sanofi Genzyme and Amicus Therapeutics and speaker’s fees from Sanofi Genzyme. UF-R has received speaker’s honoraria and unrestricted research and educational grants from Sanofi Genzyme, Shire/Takeda Pharmaceutical and Amicus Therapeutics. She is a member of Advisory Boards at Sanofi Genzyme and Amicus Therapeutics. PL has received conference registration fees and travel grants from Amicus Therapeutics and Sanofi Genzyme. MB has received salary support from Shire/Takeda Pharmaceutical. CA-B has received unrestricted investigator-initiated research grants and honoraria from Shire/Takeda Pharmaceutical and BioMarin Pharmaceutical Inc. She has service contract analyses from 4D Molecular Therapeutics, Protalix and Avrobio. She is a consultant and has received speaker’s honoraria from Amicus Therapeutics and Sanofi Genzyme. She is the Scientific director of the Waters Centre of Innovation in Sherbrooke, QC.
Provenance and peer review Not commissioned; externally peer reviewed.