Article Text

Abstract
Background Clinical interpretation of the large number of rare variants identified by high throughput sequencing (HTS) technologies is challenging. The aim of this study was to explore the clinical implications of a HTS strategy for patients with hypertrophic cardiomyopathy (HCM) using a targeted HTS methodology and workflow developed for patients with a range of inherited cardiovascular diseases. By comparing the sequencing results with published findings and with sequence data from a large-scale exome sequencing screen of UK individuals, we sought to quantify the strength of the evidence supporting causality for detected candidate variants.
Methods and results 223 unrelated patients with HCM (46±15 years at diagnosis, 74% males) were studied. In order to analyse coding, intronic and regulatory regions of 41 cardiovascular genes, we used solution-based sequence capture followed by massive parallel resequencing on Illumina GAIIx. Average read-depth in the 2.1 Mb target region was 120. Rare (frequency<0.5%) non-synonymous, loss-of-function and splice-site variants were defined as candidates. Excluding titin, we identified 152 distinct candidate variants in sarcomeric or associated genes (89 novel) in 143 patients (64%). Four sarcomeric genes (MYH7, MYBPC3, TNNI3, TNNT2) showed an excess of rare single non-synonymous single-nucleotide polymorphisms (nsSNPs) in cases compared to controls. The estimated probability that a nsSNP in these genes is pathogenic varied between 57% and near certainty depending on the location. We detected an additional 94 candidate variants (73 novel) in desmosomal, and ion-channel genes in 96 patients (43%).
Conclusions This study provides the first large-scale quantitative analysis of the prevalence of sarcomere protein gene variants in patients with HCM using HTS technology. Inclusion of other genes implicated in inherited cardiac disease identifies a large number of non-synonymous rare variants of unknown clinical significance.
- Hypertrophic Cardiomyopathy
- Genetics
- High-throughput sequencing
This is an open-access article distributed under the terms of the Creative Commons Attribution Non-commercial License, which permits use, distribution, and reproduction in any medium, provided the original work is properly cited, the use is non commercial and is otherwise in compliance with the license. See: http://creativecommons.org/licenses/by-nc/3.0/ and http://creativecommons.org/licenses/by-nc/3.0/legalcode
Statistics from Altmetric.com
Introduction
Hypertrophic cardiomyopathy (HCM), defined as left ventricular hypertrophy in the absence of abnormal loading conditions, occurs in approximately one in every 500 adults and can cause sudden cardiac death at all ages, and progressive deterioration in left ventricular function.1–6 In 50–60% of adolescents and adults with the disease, HCM is inherited as an autosomal dominant trait caused by mutations in cardiac sarcomere protein genes. Mutations in genes encoding Z-disc or calcium-handling proteins account for less than 1% of cases, and a further 5% of patients have metabolic disorders, neuromuscular disease, chromosome abnormalities and genetic malformation syndromes.7–17 The disease is characterised by a highly heterogeneous phenotype, a highly variable intra- and interfamily expressivity and incomplete penetrance. This genotype-phenotype plasticity is largely unexplained.
Although current clinical guidelines recommend routine genetic testing in patients with HCM,18 ,19 its use in everyday clinical practice has been limited by the cost and complexity of conventional sequencing technologies. Advances in high throughput sequencing technology (HTS) have the potential to solve this problem by analysing substantially larger genomic regions at a lower cost than conventional capillary Sanger sequencing,20 but they may also pose new challenges. In particular, the potential to identify a large number of rare variants that are also found in the general population, and which have little or no effect on disease phenotypes, could make attribution of causality to candidate variants using conventional methods, such as cosegregation analysis in large pedigrees, impractical.
The aim of this study was to explore the clinical implications of a HTS strategy for patients with HCM using a targeted HTS methodology and workflow developed for patients with a range of inherited cardiovascular diseases. By comparing the sequencing results with published findings, and with sequence data from a large-scale exome sequencing screen of 1287 Caucasian UK individuals (UK10K project, http://www.uk10k.org), we quantify the strength of the evidence supporting causality for detected candidate variants.
Methods
Patients and clinical evaluation
The study cohort comprised unrelated consecutively evaluated patients with HCM referred to an inherited cardiovascular disease unit at The Heart Hospital, University College London (UCL), London, UK. Patients underwent 12-lead ECG, echocardiography, symptom-limited upright exercise testing with simultaneous respiratory gas analysis and ambulatory ECG monitoring. HCM was diagnosed in probands when the maximum left ventricular wall thickness (MLVWT) on two-dimensional echocardiography measured 13 mm or more in at least one myocardial segment, or when MLVWT exceeded two SDs corrected for age, size and gender in the absence of other diseases that could explain the hypertrophy. In individuals with unequivocal disease in a first-degree relative, a diagnosis was made using extended familial criteria for HCM.21
Targeted gene enrichment and sequencing for case samples
The project was approved by the UCL/UCLH Joint Research Ethics Committee. All patients provided written informed consent and received genetic counselling prior to venesection. Blood samples were collected at routine clinic visits, and DNA was isolated from peripheral blood lymphocytes.
The study was designed to screen 2.1 Mb of genomic DNA sequence per patient, covering coding, intronic and selected regulatory regions of 20 genes known to be associated with HCM and dilated cardiomyopathy (DCM), 17 genes implicated in other inherited cardiomyopathies and arrhythmia syndromes, and a further four candidate genes (table 1).
Name of the targeted genes, Ensembl accession number, chromosomal position and size
A web-based design tool, eArray (Agilent Technologies, Santa Clara, California, USA) was employed to design an initial SureSelect (Agilent) capture library of oligonucleotides (RNA bait groups) based on the target gene sequences using the following parameters: library size 1×55 K; length 120; tiling 1×. Control samples from patients with HCM who were known to carry disease-causing sequence variants, previously detected with conventional Sanger sequencing, were used in pilot studies to validate the method. The library was used to capture the target regions from eight patients which were then sequenced (single end) on an Illumina GAIIx platform with 35 base read length. Single-end sequencing of control samples with known HCM-related variants identified regions of low coverage possibly associated with suboptimal sample processing steps or low capture efficiency. The pilot study was used to optimise the protocol and to redesign the capture library, introducing double density cover to regions of low coverage, and increase cover at the 5′ regulatory ends of genes. The following steps were adopted: single-end adapters were replaced with paired-end adapters; sequencing read length was increased from 35 bp to 75 bp, and the capture RNA bait library was redesigned with eArray to enrich regions with low coverage. The new design included an additional 965 RNA baits at 2× tiling for all <30 read depth regions, 2× tiling for 20 genes associated with HCM and DCM and redesigned sequence regions were extended 5–10 Kb upstream of all genes. As a result, 23 637 120 bp RNA baits were redesigned to target a total of 2.1 Mb of genomic DNA. In order to increase efficiency and reduce costs, we adopted a 75 bp paired-end multiplexed sequencing method which allowed us to pool 12 samples into a single lane of an Illumina GAIIX flow cell and, thus, taking into account internal controls, sequence a total of 84 samples in a single instrument run. The multiplex sequencing protocol was also tested using control samples. In early sequencing test runs a total of 21 samples from HCM patients were sequenced as part of developing and optimising the method (data not shown).
For phase two, sample preparation was carried out as recommended by Agilent and initially based on the SureSelect Target Enrichment for Illumina paired-end multiplexed sequencing method. Genomic DNA shearing (3 µg) per patient was performed on a Covaris E220 instrument in 96-well plates. Fragmented DNA was end-repaired and ‘A’ base addition was performed using the NEBNext DNA Sample Prep Master mix Set 1 (New England BioLabs). Ligation of indexing-specific paired-end adapters to DNA samples was performed using the Illumina Paired-End Genomic DNA Sample Prep Kit, and the subsequent amplification of the adapter-ligated library was carried out with Herculase II Fusion DNA Polymerase (Agilent). Hybridisation of amplified libraries to the SureSelect biotinylated RNA library (baits) was performed at 650°C for 24 h on a GeneAmp PCR System 9700 (AppliedBiosystems). Addition of index tags to the library preparation was achieved by PCR using the Illumina Multiplex Sample Preparation Oligonucleotide Kit and Herculase II Fusion DNA Polymerase (Agilent). Following the introduction of the SureSelectXT Target Enrichment protocol, all the above steps were performed with Agilent reagents as recommended by the manufacturer. Clean-up of DNA samples was performed according to the protocol using Agencourt AMPure XP beads and Dynal MyOne Streptavidin T1 magnetic beads (Invitrogen, hybridisation step). Quality of DNA samples throughout the protocol was assessed with Agilent 2100 Bioanalyzer DNA assays. All steps were performed manually using individual PCR tubes or 96-well plates except the incubation of the hybrid-capture/bead solution (hybridisation step) on 96-well plates, which was carried out on the Bravo Automated Liquid Handling Platform (Agilent) using an Agilent instrument protocol with small modifications. Samples were subjected to standard Illumina protocols for cluster generation and sequencing. Paired-end multiplexed sequencing was performed on an Illumina GAIIX with 12 samples tagged with different index sequences (Illumina) combined in each lane.
Potentially pathogenic variants and variants with a read depth below 15 were confirmed by conventional dideoxy sequencing using BigDye Terminator V.3.1 sequencing chemistry (AppliedBiosystems) on a 3130 × l capillary sequencer (AppliedBiosystems). We also sequenced, using conventional dideoxy sequencing, those exons with an average read depth below 15. All MYH6 variants were also confirmed using dideoxy sequencing, due to the high homology between MYH7 and MYH6 at DNA level, which could potentially generate false positive results.
Sequencing of UK10K control samples (http://www.uk10k.org)
DNA (1–3 μg) was sheared to 100–400 bp using a Covaris E210 or LE220 (Covaris, Woburn, Massachusetts, USA). Sheared DNA was subjected to Illumina paired-end DNA library preparation and enriched for target sequences (Agilent Technologies; Human All Exon 50 Mb ELID S02972011) according to the manufacturer's recommendations (Agilent Technologies; SureSelectXT Automated Target Enrichment for Illumina Paired-End Multiplexed Sequencing). Enriched libraries were sequenced using the HiSeq platform (Illumina) as paired-end 75 base reads according to the manufacturer's protocol.
Bioinformatic sequence data analysis
For the HCM samples, paired-end reads were aligned using the Novoalign software V.2.7.19 on the human reference genome build hg19 using quality score calibration, soft clipping and Illumina adapter trimming. Following the exclusion of PCR duplicate reads (Picard MarkDuplicate tool), insertion-deletions (indels) and single-nucleotide polymorphisms (SNPs) were called using the software SAMtools (V.0.1.18, using single sample calling).22 Variants (SNPs/indels) were filtered on the basis of the Phred scaled genotype quality score (minimum value of 30, as computed by SAMtools). For the UK10K samples, alignment was performed using Bowtie and the calling algorithm merged the output of SAMtools (V.0.1.17, single sample calling) and GATK Unified Genotyper (V.1.3-21). All samples were annotated using Annovar.23 We defined our set of candidate variants for further analysis based on frequency and function. The frequency filter used the allele frequency estimates from the 1000 genomes project database,24 and we used a 0.5% cut-off (based on the November 2010 and May 2011 releases). For the functional filter, exonic non-synonymous, loss-of-function and splice-site variants located in one of the 41 targeted genes were included in this candidate set. After filtering, variants present in the dbSNP build 135 database25 were identified, but not excluded from the analysis. We also identified variants that were previously published in the literature as disease causing mutations. Prediction of in silico pathogenicity for novel missense variants was performed using Polyphen2 and SIFT prediction software.26 ,27 A variant was predicted to be pathogenic if it was classified as ‘damaging’ by SIFT and ‘possibly or ‘probably damaging’ by Polyphen2.
Analysis of UK10K control samples
Sequencing results were compared with a set of 1287 UK controls with exome sequence data generated by the UK10K project (http://www.uk10k.org). These samples are the subset of UK10K exomes for which ethics enabled their use as control samples. None of the UK10K control samples was recruited on the basis of a cardiac phenotype. To limit the technical difficulties associated with comparing sets of variants in controls and cases generated using different protocols and analysed with the same tools but in different laboratories, we restricted our comparison to non-synonymous SNPs (nsSNPs), hence excluding indels and larger copy number variants. We retrieved the data from the UK10K project and annotated nsSNPs as candidates using the same protocol and thresholds that were applied to the set of HCM cases. To avoid biases associated with variable coverage between cases and controls, we only considered in this case-control comparison the exons that were sequenced with a read depth of 10 or more in both the UK10K dataset and our HCM case collection.
Statistical assessment of the case control comparison of candidate nsSNPs
This analysis was restricted to the 180 Caucasian HCM cases, which could be matched to the 1287 UK10K control samples. All computations were performed using the statistical software R. Frequencies of candidate nsSNPs were compared between cases and controls. We then used the case-control data to infer the proportion of HCM cases explained by rare nsSNPs variants in each gene. We used a profile likelihood approach to estimate this parameter of interest (see online supplementary material—additional statistical methods).
For genes showing a significant excess of rare nsSNPs, point estimates for the probability that a rare nsSNP is causal for HCM were estimated using the formula: (proportion of carriers of rare nsSNPs in cases—proportion of carriers of rare nsSNPs in controls)/proportion of carriers of rare nsSNPs in cases (see online supplementary material—additional statistical methods).
Results
Study population
Two-hundred-and-twenty-three unrelated patients with HCM were studied. The mean age at initial evaluation was 46±15 years (5–76); 165 (74%) were men. Mean MLVWT was 19.5±4.6 mm. Table 2 summarises the demographic and clinical characteristics of the patients.
Demographic, clinical and echocardiographic characterisation of the patients
Summary of sequence data
The median value of the per-sample average read depth in the 2.1 Mb target region across the samples was 120. Only four out 223 samples had an average read depth lower than 40, with a minimum of 20. Combining all samples and taking the median value across all samples, 91.2% of the target region was covered to a depth of 10 or more, and 85.2% to a depth of 20 or more (figure 1). Variants were filtered on the basis of the SAMtools Phred scale quality score greater than 20. We successfully validated, by Sanger sequencing, all 50 variants with a sufficient quality score but a read depth lower than 15, except for one frameshift deletion in KCNH2. This indicates that the approach has a very low false positive rate even with a limited read depth. We also screened the following genomic regions using conventional dideoxy sequencing, because of low average read depth across the samples: MYBPC3 exons 5 and 13; TNNI3 exon 1; KCNH2 exons 1 and 13. Only one false negative was found, c.459delC, p.P153fsX5 in MYBPC3. This indicates that the approach has a very low rate of false negatives. Additionally, all the MYH6 variants detected with HTS were confirmed by Sanger sequencing.
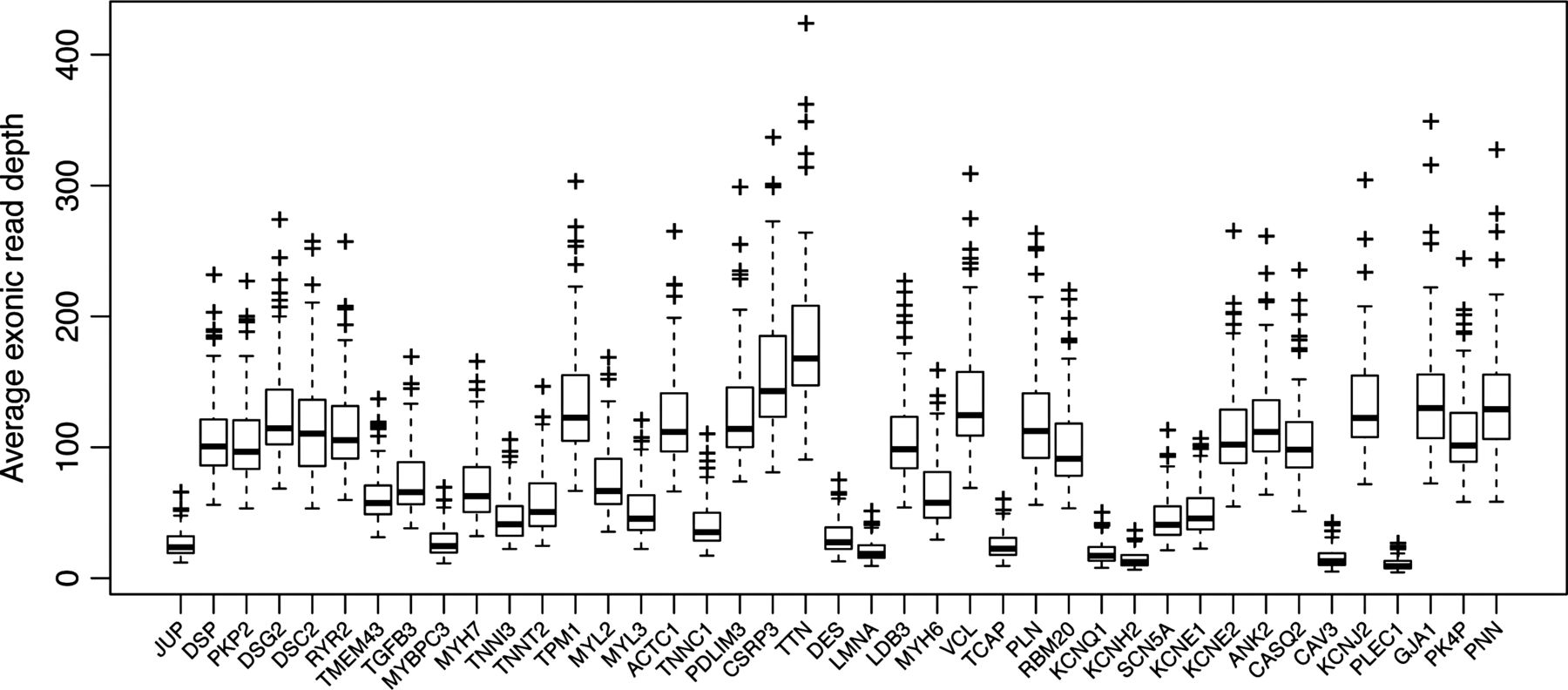
Box-and-whisker plot, showing the read-depths across the targeted genes.
Genotyping results
Initial genotype calling generated 21 939 exonic and splice-site calls distinct from the reference sequence, that corresponded to a total of 1758 distinct variants present in the 223 patients. After exclusion of synonymous substitutions, we found 9180 exonic and splice-site calls (994 distinct variants).
After filtering (as described in the methods), we selected 480 distinct rare non-synonymous exonic or splice-site variants (641 calls) as candidates for further analysis. In total, 209 patients (93.7%) carried at least one variant in the target genes, 177 when excluding TTN (79.4%). One-hundred-and-sixty-one (72.2%) patients carried multiple variants, 98 when excluding TTN (43.9%).
Variants in sarcomeric, Z-disc and calcium-handling genes
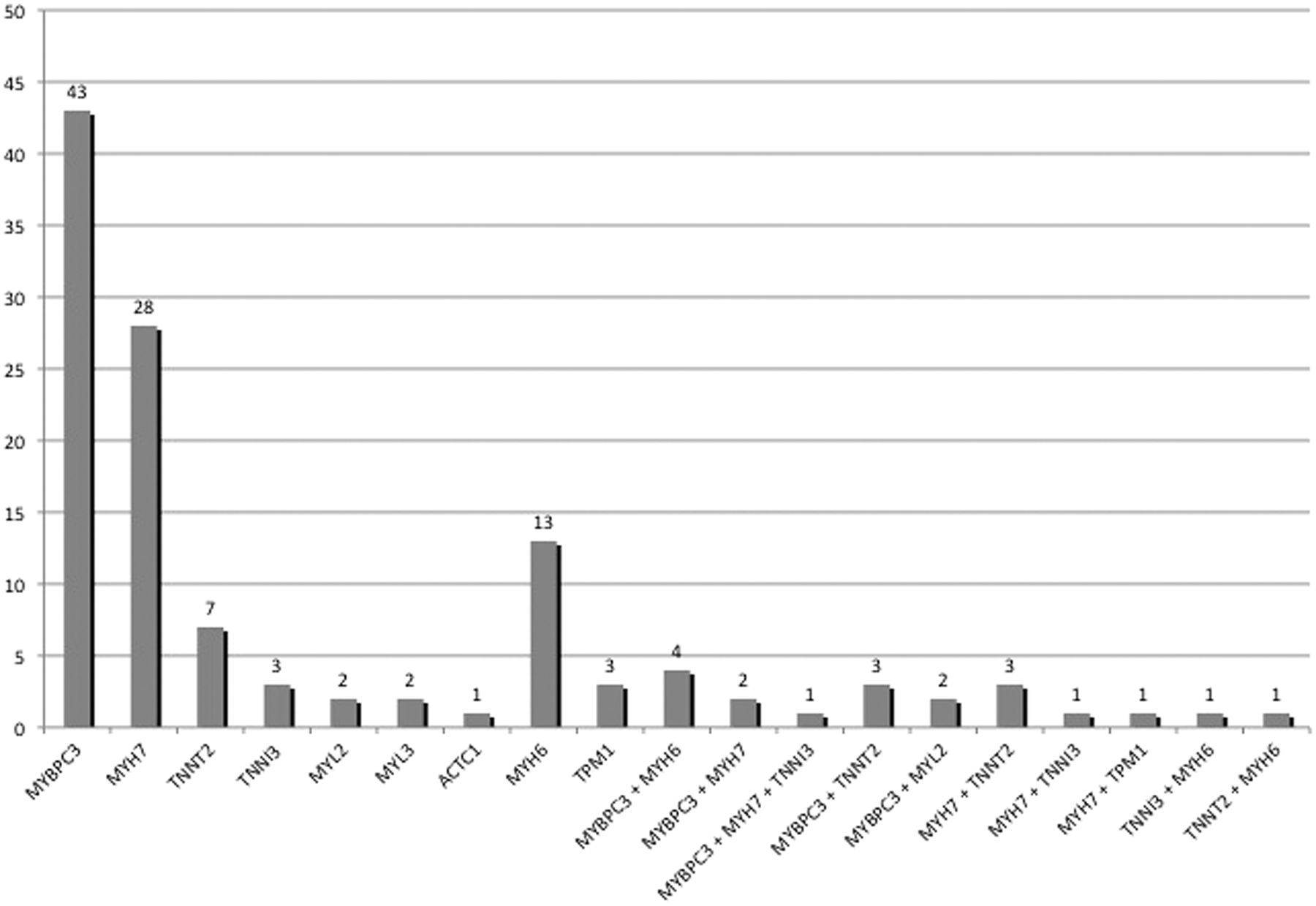
One hundred-and-two distinct rare variants in eight sarcomeric protein genes (MYH7, MYBPC3, TNNT2, TNNI3, MYL2, MYL3, ACTC1 and TPM1) were identified in 110 (49%) patients. Fifty-nine (58%) of these sarcomere protein gene variants were previously published as pathogenic mutations.8 ,11 ,15 ,16 ,28–63 Nineteen (19%) were novel missense variants predicted in silico to be pathogenic, and 19 (19%) were novel potential loss-of-function variants. In total, 97% of these sarcomere variants present in 106 patients (48% of our cohort) were considered strong disease-causing candidates.
The distribution of sarcomere variants, including MYH6, is shown in figure 2. Twenty-five patients (11%) carried multiple candidate variants in sarcomere protein genes. Twelve (63%) of the 19 double and triple heterozygotes carried a cardiac myosin binding protein C (MYBPC3) variant as one of the candidate sarcomere variants. Two patients were compound heterozygotes for MYBPC3, one patient was homozygous for a MYBPC3 variant, one patient was a compound heterozygote for β-myosin heavy chain (MYH7) and two patients were compound heterozygotes for α-myosin heavy chain (MYH6). Eighteen rare variants in MYH6, one previously published as a disease-causing mutation in a family with congenital heart disease64 and eight predicted in silico to be pathogenic, were present in 19 patients (8.5%).

Number of patients with variants in each of the sarcomeric genes.
Expanding the analysis to a panel of 19 sarcomeric and related genes, previously associated with HCM or DCM (sarcomeric, Z-disc and calcium-handling genes, excluding the highly variable titin) resulted in the detection of 152 distinct rare variants in 143 patients (64%) (figure 3). The number of variants found in each gene is summarised in table 3. From those 152 variants, 63 (41%), present in 79 patients (35%), have been previously published as pathogenic mutations. Thirty-seven (24%) were novel missense variants predicted in silico to be pathogenic, and 23 (15%) were novel nonsense, frameshift indels or splice-site variants predicted to cause loss-of-function (see online supplementary table S1 and table 3). In total, a majority (75%) of these variants are strong candidates for pathogenicity, and were present in 131 patients (59% of our cohort).
Number of distinct rare variants in sarcomeric, Z-disc and calcium-handling genes

Percentage of patients with rare variants in hypertrophic cardiomyopathy/dilated cardiomyopathy associated genes. This figure is only reproduced in colour in the online version.
Thirty-two distinct candidate variants in Z-disc and calcium-handling genes were detected. Three were previously published as disease-causing mutations: one cysteine and glycine-rich protein 3 (CSRP3) published pathogenic mutation55 (in DCM); one telethonin (TCAP) published pathogenic mutation52 (in HCM); and one RNA-binding protein 20 (RBM20) pathogenic mutation54 (in DCM). Two additional CSRP3 variants were predicted to affect a canonical splice-site, probably causing loss-of-function, and 10 novel missense variants were predicted in silico to be pathogenic: one phospholamban (PLN), one CSRP3, one lamin (LMNA), four RBM20, one vinculin (VCL), one desmin (DES) and one LIM-domain binding protein 3 (LDB3).
Table 4 shows the distribution of patients according to the strength of the evidence supporting causality for the detected candidate variants.
Statistical analysis of nsSNPs case-control data
For each gene, we combined nsSNPs to test for an overall enrichment in HCM cases compared with the general population. For our control set, we used the UK10K exome sequence dataset (Methods). To avoid technical artefacts associated with indel calling, and to properly match cases and controls, we restricted this analysis to nsSNPs and the 180 HCM Caucasian HCM cases. Data for the eight sarcomere genes most commonly implicated in HCM are summarised in table 5 and figure 4. A complete table of 19 sarcomere and associated genes is provided in online supplementary table S2. Four out of the 20 cardiomyopathy genes (MYH7, MYBPC3, TNNI3, TNNT2) showed an excess of rare nsSNP in cases compared with controls (two-tailed Fisher exact p<0.05), which is consistent with the established causal role of rare nsSNPs in these genes. We used these case-control data to extrapolate the proportion of HCM cases in the general population explained by variants in each of these genes (Methods and table 5). Rare nsSNPs in MYH7 explained the largest fraction, with between 9.6 and 20.7% of HCM cases (95% CI). We note that MYBPC3 harbours a significant number of loss-of-function indels that are excluded in this analysis, therefore, underestimating the contribution of MYBPC3.
Level of evidence for the pathogenicity of the distinct variants. (‘Others’—novel missense variants not predicted to be pathogenic in silico)
Rare nsSNPs frequency comparison between our sequencing results and a set of 1287 UK controls with exome sequence data generated by the UK10K project (http://www.uk10k.org) for the eight sarcomere genes most commonly associated with HCM

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Rare nsSNPs frequency comparison between our sequencing results and a set of 1287 UK controls with exome sequence data generated by the UK10K project (http://www.uk10k.org) for the eight sarcomere genes most commonly associated with hypertrophic cardiomyopathy. nsSNP: non-synonymous single nucleotide polymorphism. The frequency of MYH7, MYBPC3, TNNT2 and TNNI3 candidate nsSNPs is significantly higher in our cohort, as also shown in table 5. This figure is only reproduced in colour in the online version.
Assuming that an excess of candidate variants in HCM cases reflects their disease-causing potential, these data can be used to estimate the probability that a candidate nsSNP found in a HCM case is disease causing (Methods and table 5). We found that these estimates are largely dependent of the genetic variability for each gene in the general population. As an example, nsSNPs in MYH7 explain between 9.6% and 20.7% of HCM cases, and a rare nsSNP in MYH7 found in a HCM case is estimated to have 86% probability to be causal for HCM (table 5). Higher estimates are obtained for rare nsSNP in the genes TNNT2 and TNNI3, even though the contribution of these genes to HCM cases is lower. This is a consequence of the much reduced presence of nsSNPs in these genes in the control population (0.3% and 0%, respectively) compared with MYH7 (2.5%)—table 5.
Recurrent variants in HCM cases
We then investigated whether nsSNP variants were found in multiple UK HCM cases, suggesting potentially common genetic causes of HCM. We found three rare nsSNPs for which the single SNP case-control p value was less than 0.05, in MYPC3, MYH7 and TNNT2 (table 6). All were previously published as disease-causing mutations.
Candidate variants present in multiple HCM cases for which the single nsSNP case-control p value between HCM cases and UK10K controls is p<0.05
Titin
Two-hundred-and-nineteen rare titin variants were identified in 142 probands (63.6%). Two-hundred-and-nine were novel missense variants. One of the variants is a missense mutation previously described as pathogenic in HCM (R8500H).65 Nine other variants were predicted to cause loss of function in 10 patients: two are frameshift insertions potentially leading to a truncated protein (one in a patient that carried one TNNT2 variant of unknown significance, the other in a patient with a MYBPC3 splice-site variant); another is a nonsense variant that probably leads to the synthesis of a truncated protein, and six more are splice-site variants predicted to cause exon skipping (one in a patient that also carried a frameshift small deletion in MYBPC3, and another in a patient who carried a published MYH7 mutation; the other four patients did not carry any other sarcomere or related variant) (see online supplementary table S1). Thirty patients (13%) had titin candidate variants in isolation. Twenty-two patients (10%) had titin variants only in association with desmosomal gene candidate variants or ion channel disease-associated gene variants, but not other sarcomere (or related) variants. This means that 171 patients (77% of the cohort) carried a TTN candidate variant in association with sarcomere, Z-disc or calcium-handling gene variants. By contrast, in the 11 patients (5%) carrying potentially truncating variants or the published mutation, six patients carried the TTN variant isolated or only associated with ion-channel/desmosome genes (55%), and five patients in association with sarcomere variants (45%).
Variants in genes associated with arrhythmogenic cardiomyopathy and ion-channel disease
Ninety-four distinct candidate variants in genes implicated in arrhythmogenic cardiomyopathy (44) and ion-channel disease (50) were present in 96 patients (43%). Twenty-one (24%) of these variants were previously published66–81 (including 13 published variants of unknown significance in desmosomal genes, one disease-causing mutation in PKP2, and seven disease-causing mutations in 12 patients—5%—in ion-channel disease genes; table 7 and see online supplementary table S1). A further 20 variants (23%) are novel missense variants predicted in silico to be pathogenic, and three (3%) are potential loss-of-function variants. In total, aproximately half these variants are predicted to have a biological effect. Approximately two-thirds (57 patients) of these 96 patients also carried variants in sarcomere or related genes.
Number of distinct rare variants in genes associated with arrhythmogenic cardiomyopathy and ion channel disease
Discussion
This study provides the first large-scale quantitative analysis of the prevalence of sarcomere protein gene variants in patients with HCM using HTS technology. Our HTS protocol achieved adequate coverage of the targeted genomic DNA, identifying likely pathogenic sarcomeric variants in 49% of patients. We report, for the first time, the prevalence of all types of variants (including missense) in TTN and find a higher than expected number of novel rare variants in MYH6, although the total number in both genes was similar to that found in normal controls. Inclusion of other genes implicated in inherited cardiac disease resulted in the identification of a large number of non-synonymous rare variants. While the overall frequency of these variants was similar to the control population, published data and in silico prediction tools suggest that some of these have the potential to modify the disease phenotype.
Determining pathogenicity of sequence variants
Even when using conventional sequencing technology, the genetic heterogeneity of HCM and the high frequency of novel variants with uncertain effects on gene function present considerable challenges for clinical interpretation. Ideally, novel variants should be subjected to functional studies, but these are costly, time consuming, and often impractical in the clinical setting. Similarly, cosegregation analysis within families can be helpful, but is uninformative in small pedigrees and often difficult to orchestrate.
The recent availability of sequence datasets for large cohorts makes it possible to statistically compare the distribution of rare variants between controls and cases. While this statistical approach cannot on its own identify a variant as causal, it provides insights into the genetic architecture of HCM. In our study, we applied a threshold of 0.5% based on the 1000 genomes project dataset that eliminated 90% of the calls and halved the number of distinct variants, while retaining the most likely causative ones. A recently published study82 analysed the presence and frequency of DCM-associated variants in the NHLBI National Heart, Lung and Blood Institute exome sequencing project database and in the dbSNP build 131. The authors proposed a preliminary allele frequency cut-off of 0.04%. If applied to our dataset, this would exclude some clinically important variants, including five disease-causing published sarcomere variants and five published ion-channel disease-causing variants, suggesting that this threshold is too stringent.
Furthermore, we compared our sequencing results with a set of high-depth exomes generated by the UK10K sequencing project (http://www.uk10k.org) and investigated whether we could identify an excess of rare variants in cases that would be consistent with a pathogenic role. Four sarcomeric genes (MYH7, MYBPC3, TNNI3, TNNT2) showed a significant excess of rare nsSNPs compared with controls. Assuming a simple dominant model, we estimated that rare nsSNPs in these four genes explained between 12.7% and 53.2% of HCM cases (table 5). Additionally, we proposed a statistical approach that estimates the probability that a nsSNP candidate variant is causal (table 5).
Comparison with previous studies
Studies that have used conventional genetic sequencing techniques to screen patients with HCM suggest that approximately 50–60% of individuals carry mutations in one of eight cardiac sarcomere protein genes.8 ,83 This is also the approximate yield described in our centre, using conventional sequencing methodology.84 ,85 In this study, the distribution of variants among individual sarcomere protein genes and genes encoding Z-disc and calcium-handling proteins was similar to that reported previously, with the exception of MYH6 and titin, in which a higher frequency of variants was found.
In humans, cardiac myosin heavy chain exists as two isoforms, α and β, that are encoded by the tandemly arranged genes MYH6 and MYH7, respectively, situated on chromosome 14. As β-myosin heavy chain is the predominantly expressed isoform in human heart, most studies in patients with HCM have not screened the MYH6 gene, but evidence that mutations in MYH6 can cause HCM comes from a few case reports.86 MYH6 has also been recently implicated in familial atrial septal defects64 and sick sinus syndrome.87 However, in this study, the frequency of rare MYH6 variants in patients was similar to the control exome sequencing project population, questioning the importance of this gene in HCM. Further functional studies and family evaluation are currently being performed to determine the pathogenicity of the identified SNPs.
Several recent studies have focussed attention on the role of the largest protein in mammals, titin, in heart muscle disease.88–90 Titin is found in skeletal and cardiac muscle, where it forms an elastic filament bound at the N-terminus to the Z-disc and at the C-terminus to myosin and myosin-binding protein C. The inextensible A-band region of the filament consists of regular patterns of immunoglobulin-like and fibronectin repeats, whereas the I band region is composed of multiple extensible segments (or ‘spring’ elements) including PEVK, N2A (skeletal and cardiac muscle) and N2B (cardiac only). Titin is encoded by a single gene on chromosome 2 that undergoes complex differential splicing to produce isoforms with variable elastic properties. Titin has a major role in determining the mechanical properties of the heart through its effects on passive tension during myocardial stretch and restoring forces during early ventricular filling, and appears to be an important biomechanical sensor and organisational element within the sarcomere.88 Nevertheless, titin has been difficult to sequence and study due to its size, large number of isoforms and unsolved tertiary structure. In this study, we identified a large number of novel titin variants in two-thirds of the probands, the majority occurring in association with variants in other genes. Nine of the titin variants, present in 10 patients (4% of the cohort) are predicted to cause loss-of-function, which is more than the proportion of potentially truncating mutations recently reported in a subcohort of patients with HCM.89 Importantly, the majority of patients carrying truncating and the published mutation do not show any associated rare sarcomere or associated variant, which is strikingly different from the observed when considering all the titin variants. The significance of the large number of SNPs is more difficult to assess. All the individual variants present in our cohort occurred with a frequency less that 0.5% in the 1000 genomes project,24 suggesting that a proportion of them is, at the very least, modulators of the phenotype. However, the overall frequency of variants in the HCM cohort was actually lower than that seen in the control exome population. This latter finding is difficult to interpret because the annotation of titin variants is made extremely complex by the large number of possible isoforms/transcripts which are not accounted for in existing databases. Further work on understanding the role of titin in HCM is necessary.
Clinical significance of non-sarcomeric variants
Heterozygous mutations in genes encoding desmosomal proteins have been identified in up to 70% of patients with non-syndromic autosomal dominant forms of arrhythmogenic right ventricular cardiomyopathy,91 and latterly in up to 15% of patients with DCM.92 In this study, we identified a large number of desmosomal candidate variants, most of which were classified as variants of unknown significance. As with TTN, the majority occurred in patients who had at least one sarcomere protein (or related) gene variant, making it difficult to determine their pathogenic role. The same was true for the many variants detected in ion channel genes. Nonetheless, we speculate that the previously published pathogenic mutations in RYR2, ANK2, CAV3 and SCN5A may be potential phenotype modifiers in HCM, and we are now clinically re-evaluating patients with these variants.
Clinical implications
The targeted HTS protocol used in this study produced similar results to a conventional Sanger sequencing protocol focussed on a small number of sarcomere genes, but also identified large numbers of rare non-synonymous sequence variants in non-sarcomeric genes. These additional data are important as they suggest a possible role for hitherto unsuspected disease-modifying genetic variants in the disease and highlight the challenge that increasing use of HTS will pose for variant interpretation and genetic counselling in everyday clinical practice.
Standard approaches to variant interpretation will increasingly need to be complemented by other strategies and the novel quantitative methods presented in this study provide one way of determining the probability that a variant is disease causing. Additional tools that integrate genetic data with high throughput functional analyses and more sophisticated in silico prediction models coupled with improved clinical phenotyping will also be required.
Conclusions
A targeted HTS strategy in HCM identifies a large number of nsSNPs in sarcomeric and non-sarcomeric genes. Four sarcomeric genes (MYH7, MYBPC3, TNNI3, TNNT2) showed an excess of rare single non-synonymous SNPs (nsSNPs) in cases compared with controls. The frequency of non-sarcomeric variants was similar to the control population, but the clinical significance of individual variants requires further study.
Acknowledgments
We acknowledge the use of several UK10K cohorts with exome sequence data as control samples for our analysis: Edinburgh Schizophrenia Samples, Trinity College Dublin Autism Genetics Collection, Disorders of thyroid hormone synthesis and action cohort from Krishna Chatterjee (Wellcome Trust and NIHR supported), Simon Broome Register Familial Hypercholesterolaemia Samples, The Molecular Genetics of Neuromuscular Disorders Study, The Cambridge Severe Insulin Resistance Study Cohort, University College London Schizophrenia Family Samples, University College London Schizophrenia Family Samples, Edinburgh MR-psychosis samples, Scottish schizophrenia cases.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
- Data supplement 2 - Online tables
Footnotes
LRL and AZ contributed equally to this paper and are join first authors
-
Contributors All authors included on the paper fulfil the criteria of authorship. Every author contributed to all of the following: (1)conception and design, acquisition of data, and analysis and interpretation of data; (2) drafting the article or revising it critically for important intellectual content and (3) final approval of the version to be published. No one else who fulfils the criteria has not been included as an author.
-
Funding LRL is supported by a grant from the Gulbenkian Doctoral Programme for Advanced Medical Education, sponsored by Fundação Calouste Gulbenkian, Fundação Champalimaud, Ministério da Saúde e Fundação para a Ciência e Tecnologia, Portugal. CG is a British Heart Foundation (BHF) funded PhD student. VP is partly supported by the Medical Research Council (MRC) (grant G1001158). This work, including support for AZ and CD, was undertaken at UCLH/UCL, who received a proportion of funding from the Department of Health's National Institute for Health Research (NIHR) Biomedical Research Centres funding scheme. The Wellcome Trust award WT091310 supported the UK10K project.
-
Competing interests None.
-
Ethics approval UCL/UCLH Joint Research Ethics Committee.
-
Provenance and peer review Not commissioned; externally peer reviewed.
-
Open Access This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 3.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/3.0/