Article Text

Abstract
The initial mechanism for severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) infection is the binding of the virus to the membrane-bound form of ACE2, which is mainly expressed in the lung. Since the heart and the vessels also express ACE2, they both could become targets of the virus. However, at present the extent and importance of this potential involvement are unknown. Cardiac troponin levels are significantly higher in patients with more severe infections, patients admitted to intensive care units or in those who have died. In the setting of COVID-19, myocardial injury, defined by an increased troponin level, occurs especially due to non-ischaemic myocardial processes, including severe respiratory infection with hypoxia, sepsis, systemic inflammation, pulmonary thrombosis and embolism, cardiac adrenergic hyperstimulation during cytokine storm syndrome, and myocarditis. At present, there are limited reports on definite diagnosis of myocarditis caused by SARS-CoV-2 in humans and limited demonstration of the virus in the myocardium. In conclusion, although the heart and the vessels are potential targets in COVID-19, there is currently limited evidence on the direct infection of the myocardium by SARS-CoV-2. Additional pathological studies and autopsy series will be very helpful to clarify the potentiality of COVID-19 to directly infect the myocardium and cause myocarditis.
- myocarditis
- acute coronary syndromes
- pericardial effusion
- thromboembolic pulmonary vascular disease
This article is made freely available for use in accordance with BMJ’s website terms and conditions for the duration of the covid-19 pandemic or until otherwise determined by BMJ. You may use, download and print the article for any lawful, non-commercial purpose (including text and data mining) provided that all copyright notices and trade marks are retained.
https://bmj.com/coronavirus/usageStatistics from Altmetric.com
Introduction
Severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) is an unmatched challenge for the healthcare community across the world. Within a short span of 4–5 months since it was first detected, the infection has spread worldwide, with >3 500 000 confirmed cases and >240 000 deaths (with an overall mortality rate of about 6.9% based on confirmed cases) according to WHO statistics as of 6 May 2020.1
Respiratory involvement is the main clinical manifestation of COVID-19, ranging from mild influenza-like illness to severe pneumonia, and potentially lethal acute respiratory distress syndrome (ARDS). In a wide study from the Chinese Center for Disease Control and Prevention, the disease was classified as mild in 81.4%, severe in 13.9% and critical in 4.7% among 72 314 patients with COVID-19 (44 672 laboratory-confirmed, 16 186 suspected and 10 567 clinically diagnosed cases).2 Previous cardiovascular diseases (CVDs) and cardiovascular risk factors increase patients’ susceptibility to COVID-19. Moreover COVID-19 can worsen underlying CVDs and even trigger new cardiac complications.3 4
The aim of the present review is to provide an updated focus on the clinical meaning of troponin elevation in the setting of COVID-19.
COVID-19, ACE2 and target organs in humans
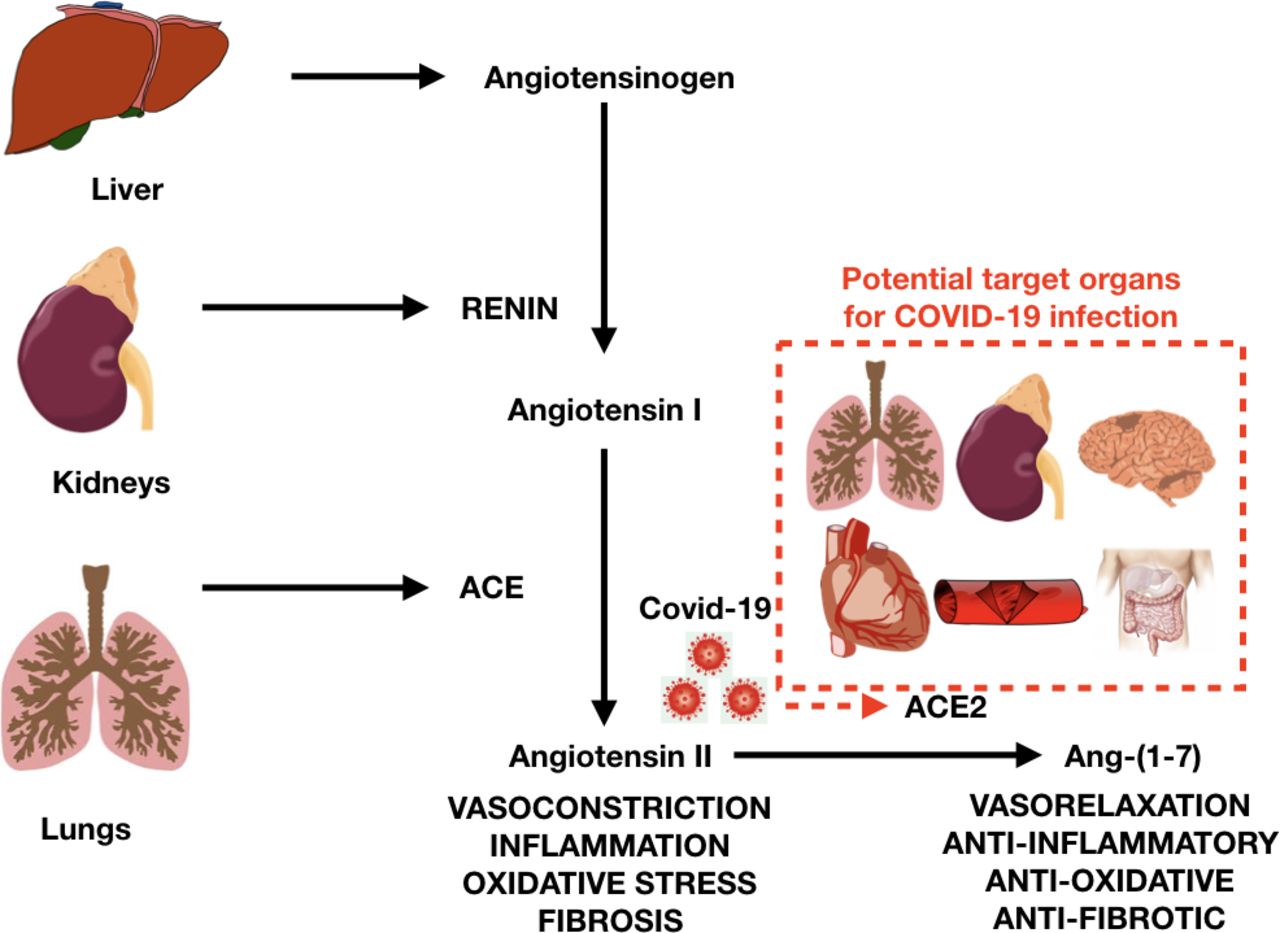
The initial mechanism for SARS-CoV-2 infection is viral binding to the membrane-bound form of ACE2 by a protein expressed in the viral coat, termed the spike (S protein),5 followed by its priming by the serine protease TMPRSS2, mediating virus uptake.6 ACE2 is a membrane-bound peptidase that is expressed in all tissues, but is especially represented in the lungs, heart, vessels, kidney, brain and gut. On this basis, all these organs are potential preferred targets of the virus, and this could explain several observations of lung, heart, vessel, brain and bowel complications, as well as symptoms and signs related to these organs during a COVID-19 infection (figure 1).7

Renin-angiotensin system, ACE and ACE2 (target of COVID-19), with potential target organs for COVID-19 infection (in red dotted line box).
ACE2 is active on peptides in the circulation, renal tubular fluid, cerebrospinal fluid, intestinal fluid and bronchial fluid. The main physiological action of ACE2 is the generation of Ang-(1-7) from Ang II. This event reduces Ang II, the major effector of the Renin–Angiotensin–Aldosterone System (RAAS), which boosts hypertension. A reduction of available ACE2 may thus increase the level of Ang II. On the contrary, ACE (not to be confused with ACE2) is responsible for the generation of Ang II and degrades Ang-(1-7). ACE2 is also responsible for the degradation of other peptides, including apelin (with cardioprotective functions) and des-arginine-1 bradykinin, which can promote inflammation.7
The internalisation of the virus with ACE2 causes the loss of ACE2 on the cell surface, causing increased levels of Ang II and decreased levels of Ang-(1-7). On this basis, the reduction in ACE2 may contribute to chronic loss of pulmonary function, inflammation and increased tissue fibrosis due to COVID-19. Since ACE2 is expressed in the heart and the vessels, they both could become targets of the virus (figure 2). However, at present the extent and importance of this compromised pathway are unknown.7 Theoretically this mechanism may help to explain why the heart can be infected and how vessels can be affected, promoting endothelial dysfunction, vascular inflammation and thrombosis, which have been anecdotally reported in advanced SARS-CoV-2 infections, as well as in patients who died of COVID-19. Moreover, these effects on the vessels, heart and hypoxia may contribute to triggering acute coronary syndromes and promote atherosclerosis. Another target organ for COVID-19 is the kidney, where ACE2 loss can be responsible for altered sodium transport, leading to increased blood volume and pressure, as well as kidney injury. The brain is another possible target, where ACE2 loss and neuronal cell deaths may cause impairment of the autonomic nervous system, regulation of blood pressure and even respiration. Since ACE2 is widespread, the cell surface diminution may contribute to a general inflammation detected in advanced cases of COVID-19.2 3
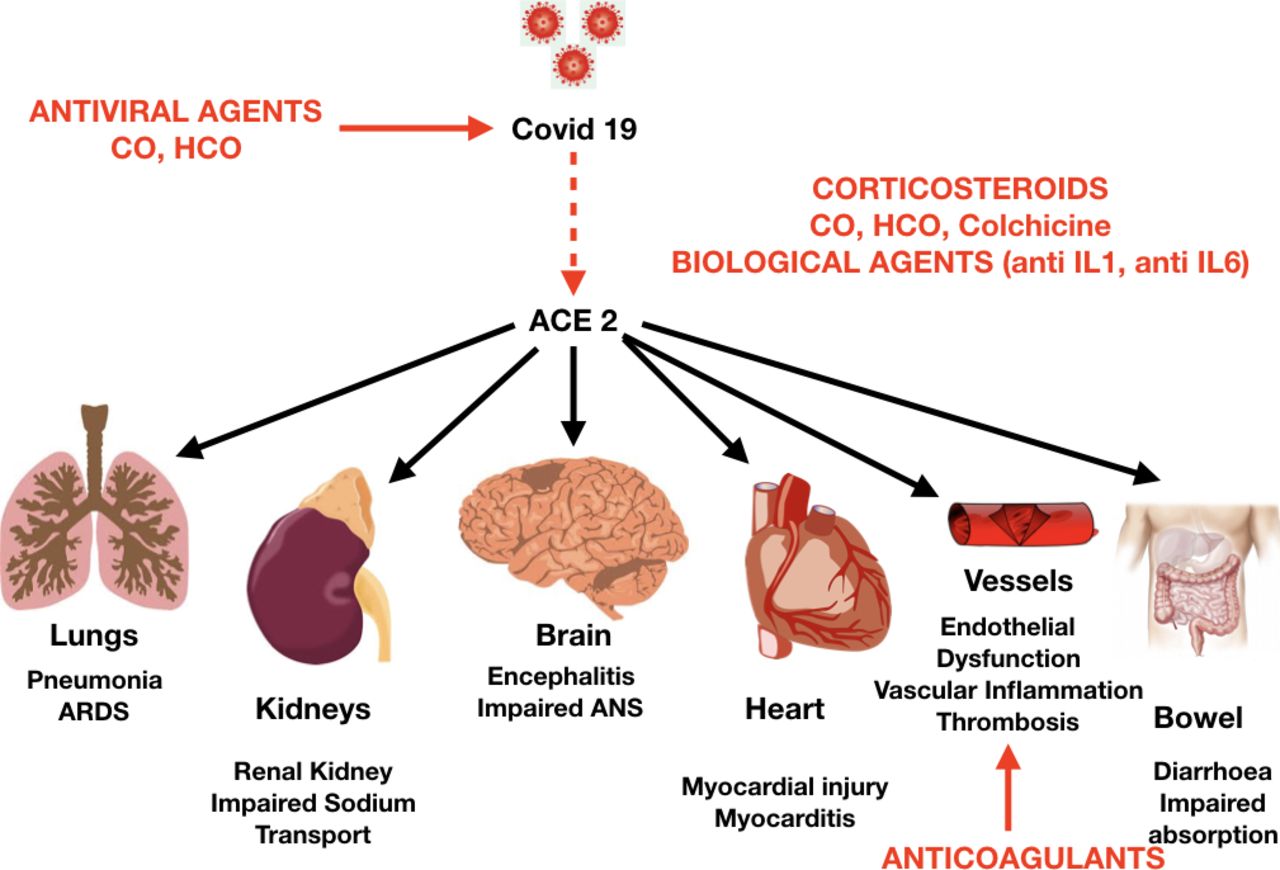
Potential target organs for COVID-19, with effects of COVID-19 infection and potential targets for medical therapy (in red). ANS, autonomous nervous system regulation; ARDS, acute respiratory distress syndrome; CO, chloroquine; HCO, hydroxychloroquine; IL, interleukin.
Experimental studies generally suggest that RAAS blockade increases ACE2 expression. Apart from RAAS blockade, exercise and statins also augment ACE2 expression.7 The influence of RAAS blockade on ACE2 has not been studied extensively, but ACE inhibitors (ACEI) and angiotensin receptor blockers (ARB) may improve the prognosis of ARDS.8
Moreover, patients with more severe COVID-19 are reported to have hypokalaemia and arterial hypertension as compared with those with milder COVID-19, suggesting a stimulation of the Ang II-AT1 receptor axis.9 Experimental evidence suggests that Ang II may promote acute lung injury and ARDS induced by coronaviruses, including SARS-CoV, SARS-CoV-2 and possibly the Middle East respiratory syndrome coronavirus.7 At present, current clinical evidence on the effects of ACEI and ARBs on COVID-19 infections is limited, and especially confined to observational studies (often of limited size) with incomplete assessment of the RAAS. On the contrary, Ang-(1-7) itself may potentially serve as a novel therapeutic to treat COVID-19. Thus, definitive conclusions cannot be drawn and targeted studies are needed, including the evaluation of statins and aldosterone blockers. ACEI do not inhibit ACE2 because ACE and ACE2 are different enzymes. Although angiotensin II type 1 receptor blockers have been shown to upregulate ACE2 in animal models, the evidence is not always accordant and varies among different angiotensin II receptor blockers. At present, no data show that ACEI or ARB promotes coronavirus entry by increasing ACE2 expression in animals and humans. Animal data suggest that elevated ACE2 expression gives protective effects. In conclusion, according to currently available evidence, ACEI and ARB should not be discontinued due to coronavirus infection.10 Moreover a recently published study confirmed that there is no increased risk of developing severe COVID-19 among >5800 patients who were COVID-19-positive and were on five common classes of antihypertensive medications (ACEI, angiotensin-receptor blockers, beta-blockers, calcium-channel blockers or thiazide diuretics).11 In an additional large, population-based study in the Lombardy region of Italy including >6200 patients, the use of ACEI and ARBs was more common among patients with COVID-19, simply due to the higher prevalence of CVD among these patients. However, there was no evidence that these drugs increased the risk of developing COVID-19.12
COVID-19, troponin elevation and myocardial injury
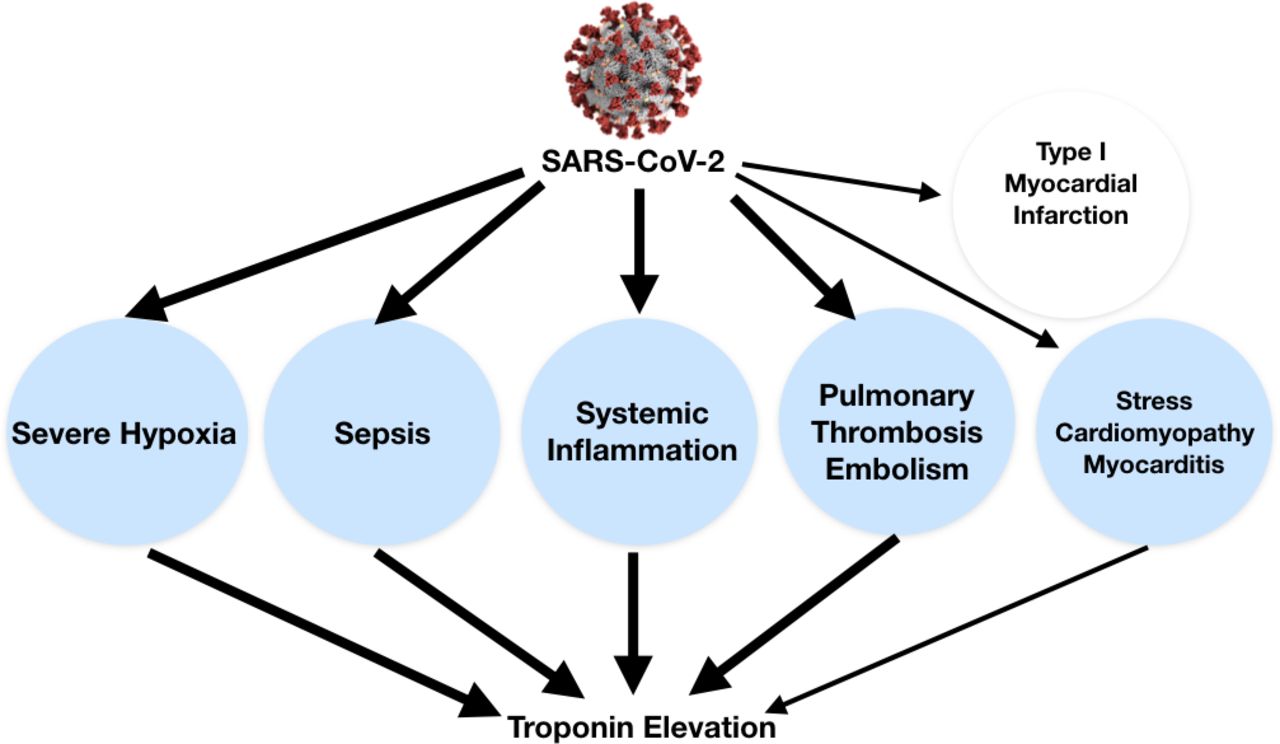
Cardiovascular tissues or generally cells that express ACE2, including lung cells, are at risk for SARS-CoV-2 infection.7 There is evidence that infection of secretory cell population in the bronchial branches is influenced not only by ACE2 expression, but also by the proteases TMPRSS2 and Furin as potential cofactors.6 So far, it is not known what role these proteases play in the binding, uptake and replication of SARS-Cov-2 in the heart cells and thus in mediating cardiac damage. In the setting of COVID-19, myocardial injury, defined by an increased troponin level, occurs especially due to non-ischaemic myocardial processes, including severe respiratory infection with hypoxia, sepsis, systemic inflammation, pulmonary thrombosis and embolism, cardiac adrenergic hyperstimulation during cytokine storm syndrome, and possibly myocarditis (figure 3). In a systematic review of four studies including 374 patients, cardiac troponin I levels were significantly higher in those with severe COVID-19 infection compared with those with non-severe disease (OR 25.6, 95% CI 6.8 to 44.5).13 Cardiac injury has been reported from 7% to 17% of hospitalised patients in China,3 14 15 and it is more common in patients admitted to intensive care units (ICU), being reported in up to 22% of cases, or in those who died (up to 59%).3 Nevertheless high-quality data and standardisation of criteria are lacking, and additional studies are warranted using the most recent universal definition of myocardial infarction.16 Moreover, in many patients troponin elevation may be increased by concomitant renal failure, which is not uncommon in advanced SARS-CoV-2 infection. Ischaemic elevation of troponin can be anticipated in COVID-19 due to combined effects on the vessels with induced prothrombotic endothelial dysfunction and systemic and vascular inflammation (figures 2 and 3).7 At present, few case reports of ST segment elevation myocardial infarction (STEMI) presenting in the context of COVID-19 have been published. In the largest report of 18 patients with COVID-19 with ST segment elevation on electrocardiography, 8 (44.4%) had a clinical diagnosis of myocardial infarction. These patients had higher median peak troponin and D-dimer levels than the 10 (56%) patients with non-coronary myocardial injury. In this setting there was high variability in presentation and high prevalence of non-obstructive disease associated with bad prognosis. Coronary angiography was performed in 50% of cases: two out of three patients of patients had obstructive coronary disease. This evidence supports the concept that myocardial injury in patients with COVID-19 could also be due to plaque rupture, coronary spasm, microthrombi, or direct endothelial or vascular injury beyond other mechanisms.17
{kind=link}
{kind=link}
{kind=link}
Troponin elevation in the setting of COVID-19 can be related to non-ischaemic myocardial injury (blue circles) by different possible mechanisms (eg, severe hypoxia, sepsis, systemic inflammation, cytokine storm, pulmonary thrombosis and thromboembolism, stress cardiomyopathy, and myocarditis). Thicker lines underline the most common causes. SARS-CoV-2, severe acute respiratory syndrome coronavirus-2.
Another emerging mechanism of troponin elevation in patients with COVID-19 is also represented by pulmonary thrombosis and arterial and venous thromboembolism affecting a variable proportion of patients in ICUs (16%–49%), causing deep vein thrombosis, pulmonary thromboembolism, ischaemic stroke, myocardial infarction and systemic arterial embolism.18–22 COVID-19 can lead to prothrombotic endothelial dysfunction, as well as systemic coagulation activation. These mechanisms are triggered and amplified by possible direct viral action and inflammatory response.21 22
COVID-19 and myocardial inflammation
Myocardial cells are a potential target of SARS-CoV-2, and myocarditis has been reported in a limited series in China, where 7% of deaths were attributed to myocardial damage with circulatory failure without a clear, definite diagnosis of myocarditis.23 Other reports have described fulminant myocarditis in the setting of high viral load, with autopsy findings of inflammatory mononuclear infiltrate in myocardial tissue, but without evidence of COVID-19 in the myocardium.24–28 In contrast to our findings in the lungs, we did not detect SARS-CoV-2 RNA in the myocardium of five patients who died due to COVID-19. We found increased numbers of macrophages in autoptic hearts, but we did not observe lymphocytic myocarditis or significant myocyte necrosis typical for other cardiotropic virus infections including enteroviruses (Klingel K et al. 2020).
The diagnosis of myocarditis cannot be based only on troponin elevation and additional evidence is often lacking. Most cases can be labelled as clinically suspected cases.29
The definitive demonstration of the presence of the virus in the myocardium is lacking. In a single case report of a 69-year-old patient with influenza-like symptoms quickly worsening to respiratory distress and cardiogenic shock, the endomyocardial biopsy demonstrated on electron microscopy viral particles in macrophages, but not in cardiomyocytes or other specific cardiac cell types.28 The patient was successfully treated with venous-arterial extracorporeal membrane oxygenation (ECMO) and mechanical ventilation. Cardiac function fully recovered in 5 days and ECMO was removed. As discussed by the authors, it cannot be excluded that macrophages detected in the heart incorporated virus particles during viraemia. In none of the publications virus replication was found in the myocardium. In a second report, a histologically confirmed case of virus-negative immune-mediated myocarditis was described in a woman who presented with an inverted Takotsubo pattern during the course of SARS-CoV-2 infection.26
In a recently published autopsy series of patients with COVID-19, endothelial cell infection has been reported in several organs, including the heart vessels, with no sign of lymphocytic myocarditis and highlighting another possible mechanism of myocardial lesion and troponin elevation.30
At present, limited data have been published on myopericardial involvement with pericardial effusion in COVID-19 infection,31 32 including a case report of cardiac tamponade in the setting of the infection with concomitant Takotsubo cardiomyopathy.31 The case was successfully managed with pericardiocentesis, colchicine, corticosteroids and hydroxychloroquine.31 However, in none of the two cases a definitive pathological demonstration of the myopericardial localisation of the virus was reported. In COVID-19, pericardial effusion could also be secondary to systemic inflammation and haemodynamic conditions instead of direct viral infection.
Conclusions
The heart and the vessels are potential targets for COVID-19; however, at present, there are no findings which provide evidence of direct infection and replication of SARS-CoV-2 in the heart cells. Table 1 summarises the current available evidence on reported cardiovascular complications of COVID-19. On this basis, troponin elevation in the setting of COVID-19 can be explained by different causes: (1) non-ischaemic myocardial injury (more commonly) related to different possible mechanisms (eg, severe hypoxia, sepsis, systemic inflammation, pulmonary thromboembolism, cytokine storm, stress cardiomyopathy) rather than a typical viral lymphocytic myocarditis; and (2) ischaemic myocardial injury with also different potential mechanisms (eg, plaque rupture, coronary spasm, microthrombi, or direct endothelial or vascular injury). In both cases troponin elevation can be exacerbated by concomitant renal failure (figure 3). In clinical practice, the differential diagnosis should take into account either ischaemic or non-ischaemic causes of troponin elevation and the potential complex pathophysiology of COVID-19. In clinically suspected STEMI, coronary angiography should be considered whenever possible, since it is now clear that acute coronary syndromes can be triggered by the infection. At the same time, if STEMI is excluded, alternative causes of troponin elevation with non-obstructive coronary arteries should be investigated. In this context, myocarditis with pseudoinfarct presentation is a differential diagnosis of myocardial infarction with non-obstructive coronary arteries.29 Whenever feasible, cardiac magnetic resonance can allow a non-invasive diagnosis of clinically suspected myocarditis, while a definite diagnosis and proof of SARS-CoV-2 infection and inflammation would require endomyocardial biopsy. In these patients, the importance of systemic inflammation, enhanced adrenergic stimulation and renal failure in more advanced cases should be recognised.
Reported cardiovascular complications in COVID-19
On this basis, additional pathological studies and autopsy series will be very helpful to clarify the potentiality of SARS-CoV-2 to directly infect the myocardium and cause myocarditis.
References
Footnotes
FGDR, YA and GMDF are joint senior authors.
Twitter @ImazioMassimo
Contributors All authors have contributed and gave final approval for the submitted manuscript.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests MI and AB have been advisory board members for SOBI and Kiniksa. YA has been an advisory board member for Kiniksa.
Patient and public involvement Patients and/or the public were not involved in the design, or conduct, or reporting, or dissemination plans of this research.
Patient consent for publication Not required.
Provenance and peer review Not commissioned; externally peer reviewed.