Article Text

Abstract
Heart failure (HF) is the leading cause of death in adults with repaired congenital heart disease (CHD). However there is currently little evidence to guide treatment strategies in this growing group of patients. Unlike the majority of HF, which is usually caused by LV systolic or diastolic dysfunction, CHD-HF is more often a consequence of RV disease, valve dysfunction, shunting or pulmonary hypertension. It is therefore not appropriate to extrapolate from the acquired HF literature and apply it to this heterogeneous population of CHD patients. Additionally, patients with CHD have been excluded from most large trials of medical or device therapy of HF, which has resulted in small retrospective and underpowered studies in the CHD population. This article critically reviews the current knowledge about CHD-HF, paying particular attention to medical therapy in different CHD populations, cardiac resynchronisation therapy and implantable cardiac defibrillators, and the challenges of heart transplantation and mechanical circulatory support in CHD patients.
- CONGENITAL HEART DISEASE
- HEART FAILURE
Statistics from Altmetric.com
Introduction
Survival into adulthood is now the expectation for even the most complex forms of congenital heart disease (CHD). This has resulted in a rapidly expanding population of adults who underwent palliative surgery for CHD in infancy or childhood. The large majority of adults with moderate or complex forms of CHD have residual anatomic and haemodynamic abnormalities that may predispose to heart failure (HF) later in life. HF is the leading cause of death in adult CHD.1 The prevalence of HF is highest in patients with complex anatomy, including single ventricle physiology, transposition of the great arteries (TGA), tetralogy of Fallot (ToF), and those with pulmonary hypertension.w1 The number of HF hospitalisations for adults with CHD has increased >80% since 1998, and CHD patients with HF have a more than threefold increased risk of death than CHD patients admitted without HF.2 w2
The diagnosis of HF in adults with CHD is challenging because many of these patients have lived their entire lives with cardiac disease and do not detect subtle changes in their exercise capacity and underreport their symptoms. At the time of symptom recognition, the extent of ventricular dysfunction and valve disease may be severe and irreversible.
The mechanisms contributing to HF in adults with CHD are unique and differ from other forms of HF. These mechanisms include chronic pressure and/or volume loading, inadequate myocardial preservation during prior surgeries, myocardial fibrosis, surgical injury to a coronary artery, pulmonary hypertension, and neurohormonal activation. The evaluation of HF symptoms in adults with CHD must be individualised based on the underlying anatomy and should include evaluation for residual shunts, baffle stenosis, valvular or conduit dysfunction, and collateral vessels which may be amenable to percutaneous or surgical interventions. Adults with CHD are commonly excluded from clinical trials and there are few data to guide therapy in this growing population. Therefore, one must be very careful when extrapolating from general HF literature, which in large part applies to patients with LV systolic dysfunction. This article focuses on the current medical, electrophysiological, and transplantation treatment options for CHD patients with HF.
Medical therapy
The role of medical therapies, which are commonly used in non-CHD HF, has not been well established in patients with CHD. Because CHD-HF patients have levels of functional impairment and neurohormonal activation similar to those with non-CHD HF, investigators have attempted to test the effectiveness of standard HF therapies in CHD patients.w3 w4 Additionally, patients with CHD have adverse cardiac remodelling so medications such as β-blockers and ACE inhibitors may be useful. However, as discussed above, CHD-HF is less likely to be caused by LV systolic dysfunction and is more commonly due to RV dysfunction, tricuspid valve regurgitation, pulmonary hypertension or diastolic dysfunction—all areas in which HF medication has not been proven effective. Additionally, because event rates are lower in CHD patients than in non-CHD HF and sample sizes are limited, most studies have been underpowered and conclusive clinical trials in CHD-HF are rare.
Tetralogy of Fallot
As patients with repaired ToF age they are at increased risk for HF; however, no medical therapy has been proven to benefit this group of patients. A randomised placebo controlled trial of bisoprolol failed to show improvement in New York Heart Association (NYHA) functional class, peak oxygen consumption, or RVEF in 33 patients with repaired ToF.3 Ramipril (versus placebo) failed to show improvement in cardiac magnetic resonance derived RVEF in a randomised controlled trial of 64 patients with repaired ToF.4 Both of these trials were limited to a 6-month follow-up, and outcomes focused primarily on RV function, which may not have a predictable or consistent response to HF medical therapy.
LV dysfunction is being increasingly recognised as a major determinant of outcomes in patients with repaired ToF. In fact, at least 20% of these patients develop LV systolic dysfunction,5 which is associated with higher arrhythmia burden and worse outcomes.6 However, in small trials neither β-blockers3 nor ACE inhibitors4 improved LVEF in patients with repaired ToF. This may be because LV dysfunction may be due to interventricular interactions in patients with chronic RV volume overload, which might not be helped by traditional HF medications, and is possibly why LVEF appears to improve after pulmonary valve replacement.w5
Systemic RV
Another group of CHD patients at risk for HF are those with a systemic RV. The majority of these patients have either d-loop TGA and have undergone an atrial switch procedure or physiologically corrected TGA. These patients may have very different responses to medical therapy. In addition to having RV systolic dysfunction, patients with d-loop TGA who have undergone an atrial switch procedure are often limited by a fixed stroke volume through stiff atrial baffles and sinus node dysfunction—neither of which benefits from traditional HF medications (and may be exacerbated by β-blockade). Patients with physiologically corrected TGA often have primary systemic atrioventricular valve regurgitation and are at high risk for progressive atrioventricular block which also does not respond to HF medications.
There have been several small case series examining the effectiveness of β-blockers in patients with systemic RVs, with some evidence for beneficial effects on ventricular size and function.7
Data on the use of agents to inhibit the renin–angiotensin–aldosterone system in patients with systemic RVs are also limited. Therrien reported the results of a prospective, double blind, randomised, placebo controlled clinical trial of ramipril for 1 year in 17 adults with TGA who had undergone an atrial switch procedure and found no significant increase in the systemic RVEF.w6 Small trials of angiotensin receptor blockers have also shown conflicting results.8 In a multicentre, double blind, randomised controlled trial comparing valsartan to placebo in patients with a systemic RV, there was no significant effect of valsartan on RVEF, exercise capacity, or quality of life during a 3-year follow-up.9 Similar to the negative medication trials in patients with repaired ToF, one must question the endpoints chosen in these trials.w3
Fontan patients
Patients with single ventricle physiology palliated with the Fontan operation are at high risk for HF, either from ventricular dysfunction or chronic venous congestion. Fontan patients have high afterload, stroke work, and adverse ventricular–arterial coupling, and are at increased risk for ventricular remodelling and dysfunction.w7 Afterload reduction is therefore tempting but there have been no adequately sized clinical studies of afterload reduction in adults with Fontan circulation so their use is purely speculative. Additionally, as Fontan patients age, they are at risk for hepatic dysfunction which may lower the systemic vascular resistance, and afterload reduction may not be effective. Patients with venous congestion should be treated with diuretics.
Patients with Fontan circulation lack a sub-pulmonic ventricle, and low pulmonary vascular resistance is required to maintain adequate cardiac output. Pulmonary vasodilators have been tested in small studies. Acutely, sildenafil improved maximum oxygen uptake and, when taken chronically, had beneficial effects on ventilatory efficiency and anaerobic threshold in some patients.10 ,11
In summary, larger trials with newly defined clinical endpoints are needed to better define the role of medical therapy in these patients.
Transplantation and mechanical circulatory support
Transplantation
Consideration of heart transplantation is appropriate for patients with CHD who have refractory severe HF despite optimal medical and surgical management and for those who have significantly increased risk of death. However, selecting the appropriate CHD patients to list for transplantation remains challenging because, unlike acquired heart disease, there are few models to predict transplant-free survival in CHD patients. However, risk models are now being tested in CHD patients and may aid in selection in the future.w8 In some congenital populations, reduced exercise performance has been associated with poor survival and may prompt transplant listing,w9 but this finding has not been consistently demonstrated across all diagnoses. For these reasons, many adults with CHD who may benefit from transplantation are not listed in a timely fashion. A multidisciplinary team comprising CHD trained cardiologists and surgeons, as well as cardiologists with expertise in advanced HF, should evaluate each patient and consider possible medical, surgical or transcatheter treatment options before listing for transplantation.
There are often barriers to transplantation listing in CHD patients. Advanced HF patients with CHD often have complex anatomy including abnormal position of organs or vessels (eg, situs inversus, malposition of the great arteries or venous anomalies), which increases the technical difficulty of transplantation. In these cases, transplantation should be performed by a surgeon with expertise in CHD. Most CHD patients have had prior chest surgery, and repeat sternotomy may be associated with prolonged dissection and lengthy organ ischaemic time. Some adults with CHD have elevated pulmonary vascular resistance due to shunt physiology, distorted pulmonary artery anatomy and pulmonary venous hypertension, and careful haemodynamic evaluation by a congenital invasive cardiologist is required to evaluate pulmonary vascular resistance and reactivity before transplant listing.
Alloimmunisation may occur after blood transfusion, pregnancies or homograft insertions. Antibodies can persist for more than a decade after CHD surgery.12 Elevated human leucocyte antigen (HLA) antibodies can lead to organ rejection, reduce post-transplant survival,w10 and can make finding a suitable organ difficult, contributing to longer waiting times. Additionally, the presence of hepatic dysfunction and pulmonary hypertension, both of which are common in CHD patients with HF, may be contraindications to transplantation. Due to these associated findings, the majority of adults with CHD referred for transplantation evaluation do not undergo listing.13
Once listed, CHD patients are more likely to be listed at lower urgency (64% of CHD patients are listed as status 2 compared to 44% of non-CHD patients), despite a higher rate of complications once on the transplant list.14 This discrepancy may, in part, be explained by lower utilisation of mechanical circulatory support (MCS) among CHD patients that lowers urgency status. Additionally, once CHD patients achieve status 1, waiting time is longer than for non-CHD patients. For these reasons, once listed, patients with CHD are less likely to undergo transplantation (figure 1) than patients without CHD (54% vs 63%).15

Competing risks analysis of outcomes after listing for cardiac transplantation among two groups: (1) patients with CHD (dotted lines); and (2) patients without CHD (solid lines). Data are from United Network for Organ Sharing between 1995 and 2009, n=41 849; 2.5% with congenital heart disease. CHD, congenital heart disease; removed, removed from transplant list. Reproduced with permission from Davies et al.15
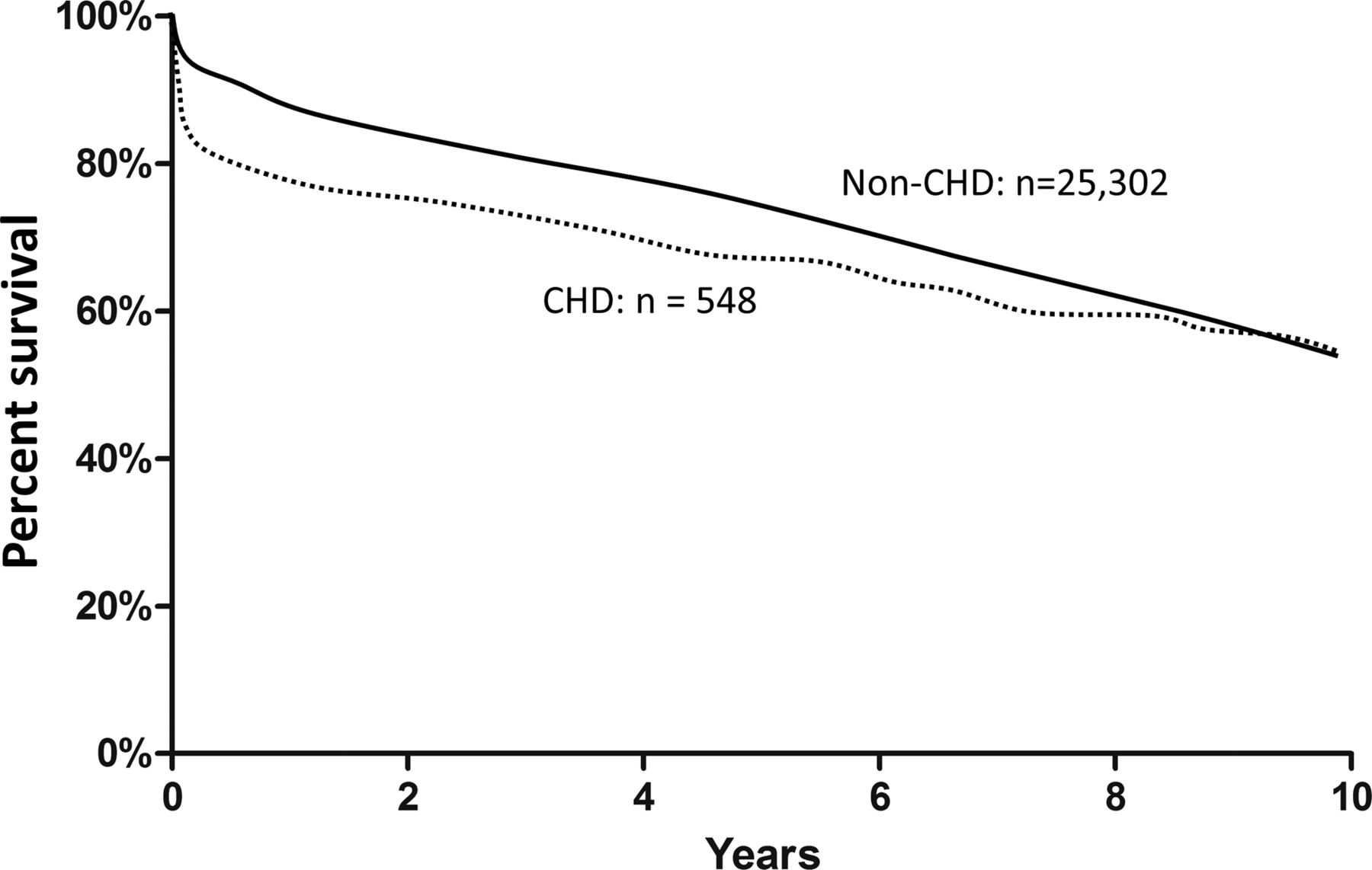
Early post-transplant mortality is increased in patients with CHD (OR 3.6) as are post-transplant complications including dialysis and prolonged intensive care unit stay. The most frequent causes of early post-transplant mortality in patients with CHD are haemorrhage and acute graft failure.w11 Nonetheless, 10-year survival is similar between CHD and non-CHD populations (figure 2).15 Risk factors for adverse transplant outcomes in CHD patients include prior Fontan operation, complex anatomy, reoperative sternotomy, and pulmonary hypertension.16 Fontan patients have higher perioperative risk as they are likely to have had multiple prior surgeries and multiorgan involvement, including congestive hepatopathy with cirrhosis, renal dysfunction, and coagulation abnormalities. Five-year survival (68%) is marginally worse for Fontan patients than for other forms of CHD.17 Liver dysfunction is often a barrier to transplant in Fontan patients; however, emerging literature suggests that the histologic finding of cirrhosis may not preclude transplantation in Fontan patients.18

{kind=link}
{kind=link}
Kaplan-Meier cumulative survival after transplantation. Ten-year survival estimates are illustrated stratified by the presence of CHD. nCHD indicates non-CHD group. Data are from United Network for Organ Sharing between 1995 and 2009, n=41 849; 2.5% with congenital heart disease. CHD, congenital heart disease. Reproduced with permission from Davies et al.15
Mechanical circulatory support
Ventricular assist devices (VADs) are not widely used in patients with CHD, although their use has increased in the past decade. The utilisation of VADs remains low in the CHD population listed for transplant (7.7%) compared with patients with acquired heart disease (>19%).15 w12 Importantly, CHD patients who have VADs before transplantation have post-transplant survival similar to CHD patients without VADs.w12
Other than administrative databases, reports of VAD use in CHD are limited to small case series of carefully selected patients. Therefore, it is difficult to generalise applicability or outcomes to a broader CHD population. VADs have been used successfully in both morphologic left and right systemic ventricles as well as in patients following the Fontan operation. HF patients with CHD are more likely to have right-sided HF, pulmonary hypertension or residual shunts, which may make them less attractive candidates for VADs. The relative unavailability of long term MCS in CHD, and the long wait time once listed, highlights the need for early referral for transplantation in CHD patients. It is important to recognise that adults with CHD may not recognise their functional limitations, and NHYA functional class is a poor predictor of exercise capacity.19 Objective exercise testing can be very helpful in this population. Additionally, a common error in referral patterns is to wait until there is ventricular systolic dysfunction, and by that time the multiorgan involvement that is present in CHD patients may preclude transplant candidacy.
Arrhythmia and cardiac resynchronisation therapy in adult CHD
The lifetime cumulative risk of arrhythmia is >50% for patients born with CHD, and arrhythmias complicate at least half of all HF admissions among patients with CHD.20 w13 A comprehensive discussion of medical, catheter based, and surgical therapy for atrial and ventricular arrhythmias in adults with CHD is an essential component in the management of HF in these patients.w14
Unlike patients with acquired heart disease, arrhythmias in CHD are often a consequence of prior surgical intervention. Sinus node dysfunction is common in patients who have undergone atrial baffle procedures (Mustard or Senning) for d-loop TGA. Atrioventricular conduction abnormalities are frequently seen following ventricular septal defect repair, aortic valve surgery or the Konno procedure for enlargement of a hypoplastic LV outflow tract.
Tachyarrhythmias are typically macro re-entrant around an anatomic obstacle such as a ventricular septal defect patch, atrial baffle, atriotomy or ventriculotomy scars. Atrial arrhythmias often appear atypical on a 12-lead ECG in patients with CHD. Atrial re-entrant tachyarrhythmias are often scar-based and the cycle length is usually long (approximately 300–500 ms). This may predispose to 1:1 ventricular conduction which has been linked with sudden cardiac death (SCD) in patients with d-loop TGA; small studies suggest that ß-blockers may be protective.21
Patients with single ventricle anatomy who have undergone a Fontan operation are at high risk for intra-atrial re-entrant tachycardia. Cycle length is typically approximately 300 ms allowing for 2:1 conduction at a ventricular rate of approximately 100 beats/min, rather than the typical 150 beats/min in isthmus dependent atrial flutter, which can make detection of this arrhythmia difficult. Arrhythmias are often precipitated by haemodynamic abnormalities, thus they are more common in Fontan patients with HF and in those with older styles of Fontan surgeries (eg, atriopulmonary Fontan, typically completed before 1987). Non-sinus rhythm is poorly tolerated and can precipitate worsening of HF in Fontan patients; therefore, restoration of sinus rhythm is preferred.w15 Stagnant flow in the Fontan circuit is common and anticoagulation is indicated for thromboprophylaxis in Fontan patients with atrial arrhythmias.22 Antiarrhythmic medications are often inadequate to control atrial arrhythmias and can worsen sinus node dysfunction that is common in Fontan patients. Radiofrequency ablation has been used successfully with high acute procedural success. However, most patients have multiple re-entrant circuits and high rates of recurrence—up to 85% at 4 years.w16 w17 When atrial arrhythmias are detected, haemodynamic perturbations should be evaluated and, when possible, treated aggressively. There should be consideration of surgical therapy with Fontan revision with a Cox-Maze procedure tailored to the Fontan anatomy.w18
Cardiac resynchronisation
Patients with chronic systolic HF and a wide QRS benefit from cardiac resynchronisation therapy (CRT). However, it is difficult to extrapolate the data from acquired heart disease to CHD because of fundamental differences in the pathophysiology of HF in CHD. Furthermore, due to persistent shunts, patients with CHD and HF are more likely to require epicardial pacing systems, which increases the morbidity of implantation and may offset the benefits of resynchronisation.
Three retrospective studies of CRT in heterogeneous CHD populations have been published (table 1).23–25 Epicardial device placement was common (up to 50%). Clinical response was not uniformly defined and included improvement in EF or functional class. Using these definitions, 32–87% of patients had improvement with CRT.
Summary of studies examining cardiac resynchronisation therapy response in pediatric and congenital heart disease patients
Due to the small number of patients studied and the heterogeneous CHD population, there are insufficient data to determine which anatomic subpopulations derive the greatest benefit from CRT. For example, the benefit of CRT in patients with systemic RVs has not been readily established.23 ,25 However, one multicentre study did show an average improvement in EF of 13% and clinical response in 13/17 patients studied.24 Other small case series have also been encouraging.26 However, given the very small number of patients studied, CRT in patients with HF and systemic RVs cannot be routinely recommended.
Implantable cardioverter defibrillators in CHD
Implantable cardioverter defibrillators (ICDs) are appropriate for secondary prevention in patients who have survived a cardiac arrest or who have experienced sustained symptomatic ventricular tachycardia.22 The use of ICDs for primary prevention in CHD patients remains controversial. Patients with single ventricle anatomy or residual shunts require epicardial leads that increase the morbidity of implantation. Additionally, CHD patients who require ICDs are likely to be young; therefore they may need multiple generator replacements, lead revisions, and accumulate more abandoned leads over a lifetime. Furthermore, CHD patients are likely to have supraventricular arrhythmias, which may account for the high rate (15–41%) of inappropriate shocks (table 2).
Outcomes of implantable cardioverter defibrillators in adult patients with congenital heart disease
Summary
HF is increasingly recognised in adults with CHD, and HF related complications are the most common cause of death in this population. Potential HF therapies include medical treatment, device therapies and surgical interventions, such as mechanical assist devices and transplantation. However, CHD-HF is fundamentally different from acquired HF and established HF therapy is often not applicable to the patient with CHD due to disease heterogeneity, unusual mechanisms of HF, and unique comorbidities in adults with CHD. Early recognition of HF in patients with CHD is essential for appropriate management as multiorgan dysfunction may precede systolic dysfunction. A multidisciplinary team with expertise in CHD and advanced HF is paramount in managing this challenging population, and multicentre trials are needed to better define the appropriate therapies for this growing group of patients.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online references
Footnotes
-
Contributors EVK and AMV both contributed to the authorship and critical review of the manuscript.
-
Competing interests None.
-
Provenance and peer review Commissioned; internally peer reviewed.