Article Text

Abstract
Objective The objective of this study was to elucidate the natural history of late-onset transthyretin Val30Met-associated familial amyloid polyneuropathy (FAP ATTR Val30Met) in non-endemic areas.
Methods The authors retrospectively assessed the development of major clinical landmarks and abnormalities of nerve conduction and cardiac examination indices in 50 patients with an age of onset older than 50 years and no relationship to endemic foci.
Results Once the neuropathic process was initiated, sensory and motor symptoms of both the upper and lower extremities appeared within a period of one and a half years. Digestive and orthostatic symptoms also tended to occur in the early phase of the disease, whereas urinary symptoms appeared in the middle of the disease progress. Along with pain in the extremities, these symptoms progressed over time and significantly disturbed the quality of life during the late phase of the disease, resulting in the need for wheelchair use. Although cardiomyopathy became clinically apparent only in the late phase of the disease, it was found to be the major cause of death. The mean duration of the disease onset to death was 7.3 years. Although values at the time of diagnosis were extremely variable, serial measurements of electrophysiological indices, the cardiothoracic ratio and interventricular septum thickness indicated a steady exacerbation in these outcomes among patients within a span of a couple of years.
Conclusions The ages of onset of each clinical landmark were extremely variable between patients. However, once an initial symptom appeared, the chronological sequence of other clinical landmarks tended to be uniform, occurring within a relatively short time span.
- Amyloid
- amyloidosis
- familial amyloid polyeuropathy
- natural history
- transthyretin
- neuropathy
- GBS
- peripheral neuropath
- motor neuron disease
Statistics from Altmetric.com
- Amyloid
- amyloidosis
- familial amyloid polyeuropathy
- natural history
- transthyretin
- neuropathy
- GBS
- peripheral neuropath
- motor neuron disease
Introduction
Transthyretin (TTR) Val30Met-associated familial amyloid polyneuropathy (FAP ATTR Val30Met) is the most common form of FAP. The condition is caused by an amino acid substitution at position 30 of TTR of valine with methionine.1–12 Although patients with FAP ATTR Val30Met are generally considered to be concentrated in geographically restricted areas of Japan, Portugal and Sweden,1–4 6 7 11 recent analyses have revealed a late-onset form of this type of FAP distributed outside these endemic areas.5 11–13 The penetrance rate of FAP ATTR Val30Met is reported to differ among various regions.4 11 12 14 15 Recent epidemiological studies suggest the presence of new endemic foci in Ishikawa Prefecture in Japan as well as in the Mediterranean country of Cyprus.14 16 These observations suggest that FAP ATTR Val30Met may not be as rare as was previously thought.9 17 18 In fact, the existence of a global population of undiagnosed patients with FAP ATTR Val30Met cannot be ruled out.11 18 Therefore, a diagnosis of neuropathy with an undetermined aetiology, particularly when occurring among the older people, should prompt the clinical consideration of FAP ATTR Val30Met.18
Although liver transplantation is an established treatment for FAP,19 patients with the late-onset form of FAP ATTR Val30Met may be too old to undergo such treatment. However, recent advances in alternative therapeutic strategies, including the prevention of amyloid fibril formation, reduction of TTR deposition by immunisation or gene therapy, have increased the value of early FAP ATTR Val30Met diagnosis.20–24 Additionally, drugs that may ameliorate the clinical course of FAP, such as tafamidis, may become available in the foreseeable future.21 In order to prepare for such a novel therapeutic approach, the natural history of FAP must first be established. Indeed, the long-term evaluation of the natural history of this disease in a significant sample of patients will become increasingly challenging as these novel therapies may modify the course of FAP.
Given that over 100 different mutations in the TTR gene have been reported, extensive phenotypic variation should be taken into consideration when assessing the natural history of FAP.6–8 11 Even patients with specific mutation, such as the Val30Met mutation, show variable clinicopathological features depending on the age of onset and geographical location.11 12 25 Such findings suggest that the evaluation of natural disease history should be performed within respective phenotypic groups. A recent study described the rate of progression of FAP over a period of 1 year in nine patients with various TTR mutations.26 However, clinical characteristics of patients with late-onset form of FAP ATTR Val30Met in non-endemic areas have only been assessed by cross-sectional studies.12 17 25 27 28 Clearly, there is a need to assess features of this condition in a longitudinal fashion.
In the current study, we sought to elucidate the natural history of late-onset FAP ATTR Val30Met in non-endemic areas. Specifically, we retrospectively assessed the development of major clinical landmarks and abnormalities of nerve conduction indices and cardiac examinations in 50 patients with this type of FAP in Japan.
Patients and methods
Patients and clinical evaluations
We retrospectively investigated 50 patients with late-onset FAP ATTR Val30Met who originated from 48 families in non-endemic areas of Japan. Inclusion criteria included a diagnosis of FAP ATTR Val30Met with an age of onset older than 50 years, as well as no relationship to the conventional endemic foci (Arao and Ogawa) or to the recently reported endemic focus (Ishikawa Prefecture)16 within two prior generations. A previous nationwide survey of FAP ATTR Val30Met in Japan revealed that a small number of early-onset (younger than 50 years) patients were present in non-endemic areas.12 Nevertheless, we did not include such patients in the current analysis as their clinical features were similar to those of patients in the endemic foci.12 Amyloid deposition was detected in all patients at the following sites: sural nerve (n=38), gastrointestinal mucosa (n=9), endocardium (n=1), eye (n=1) and bladder (n=1). Systemic amyloid deposition was confirmed at autopsy in eight patients. In order to confirm the diagnosis of FAP ATTR Val30Met, DNA analyses for mutation of the TTR gene were performed in all patients as described previously.29 30 During the observation period, none of the patients received remedies that could ameliorate amyloid deposition, such as diflunisal, tafamidis, Cr3+, carvedilol, doxycycline and tauroursodeoxycholic acid or its analogues.20 21 23 24 For one patient who received liver transplantation at age 62, only information collected before liver transplantation was included in this study. Informed consent was obtained from all patients. The study protocol was approved by the ethics committee of Nagoya University Graduate School of Medicine.
All patients underwent clinical and neurological assessment, routine blood and urine studies and cerebrospinal fluid analysis. The clinical course of the disease was assessed using the initial symptoms associated with amyloidosis and the age of onset of the following 11 clinical landmarks: sensory symptoms in the lower limbs (numbness or pain in the feet), sensory symptoms in the upper limbs (numbness or pain in the hands), motor symptoms in the lower limbs (weakness in the lower limbs), motor symptoms in the upper limbs (weakness in the upper limbs), digestive symptoms (diarrhoea or constipation defined by absence of bowel movements for more than 3 days), orthostatic symptoms (orthostatic intolerance, such as lightheadedness or subsequent syncope), urinary symptoms (nocturnal or diurnal urinary frequency, a sensation of urgency, urinary incontinence or voiding difficulty and retention), cardiac symptoms or signs (dyspnoea or oedema in the lower limbs), use of a cane (constant use of a cane when away from home), use of a wheelchair (constant use of a wheelchair when away from home) and death. The cause of death was also investigated. We assessed the age at which these respective clinical landmarks first occurred through the use of direct interviews, patient examination, family interviews and by review of the patient's clinical record. Family members were helpful in confirming the age of onset of clinical landmarks that they could easily have recognised, such as the use of a cane or wheelchair or the time of death.
Each evaluator used the same criteria to assess the onset of a given landmark. To verify concordance between different evaluators' assessments, two neurologists independently assessed the age of onset of these clinical landmarks in the progression of FAP ATTR Val30Met in 20 patients. The assessments by the two evaluators were highly concordant, as evidenced by a Pearson's correlation coefficient above 0.97 for each clinical landmark.
Previous studies investigating FAP have specified various clinical expressions of autonomic dysfunction, including orthostatic intolerance, nausea/vomiting, diarrhoea/constipation, dysuria and erectile dysfunction.7 31 32 Due to the low occurrence of nausea or vomiting in our patients with late-onset FAP ATTR Val30Met, these symptoms were not interpreted as a clinical landmark. Additionally, due to the advanced age of our patients, and the infrequency of complaint of the symptom, erectile dysfunction was not considered a clinical landmark in the present study.
Electrophysiological assessment
Motor and sensory conduction were measured using standard methods with the application of surface electrodes for stimulation and recording.17 28 33 Motor conduction of the median, ulnar and tibial nerves was investigated using a recording from the abductor pollicis brevis, abductor digiti minimi and abductor hallucis brevis muscles, respectively. The following nerve segments were used to calculate motor nerve conduction velocity (MCV): wrist to elbow for the median nerve, wrist to distally at the elbow for the ulnar nerve and ankle to popliteal fossa for the tibial nerve. Sensory conduction of the median and ulnar nerves was investigated using antidromic recording from ring electrodes at the second and fifth digit, respectively, while that of the sural nerve was investigated using bar electrodes at the ankle. Sensory nerve conduction velocity was calculated for the distal segment. Amplitudes of compound muscle action potential (CMAP) and sensory nerve action potential (SNAP) were measured from the baseline to the first negative peak.
Cardiac assessment
We investigated various cardiac findings that were suggestive of amyloid deposition.18 34 35 The presence of cardiomegaly was assessed using a chest x-ray. Two-dimensional and M-mode echocardiograpy were used to measure the interventricular septum thickness. Views of the heart were obtained from the parasternal long axis position.35 The interventricular septum thickness was measured at the end of diastole, as indicated by the Q-wave on ECG.35 Granular sparkling echo findings were defined as distinct and bright echoes that could be visualised from different projections or angulations and persisted at a gain setting low enough to eliminate echoes from the surrounding endocardium and myocardium.18 34 Serum N-terminal-pro B-type natriuretic peptide (NT-proBNP) levels were examined using an electrochemiluminescence immunoassay. Finally, a 12-lead ECG and 24-h holter ECG were performed to detect an atrioventricular conduction block.
Statistical analyses
Quantitative data were presented as the mean±SD. The Mann–Whitney U test, Kruskal–Wallis test or Pearson's correlation coefficient analyses were conducted as appropriate. A p value of <0.05 was considered to indicate statistical significance.
Results
Clinical characteristics elucidated during the follow-up period
Patients' backgrounds and clinical characteristics elucidated during the follow-up period are described in table 1. Briefly, the age of onset ranged from 51 to 77 years (64.5±6.5 years), while the age at diagnosis of the disease was 67.3±5.9 years. Patients were followed up until 70.7±6.3 years of age. Of the 48 families included in this study, 17 were found to have more than one family member diagnosed with FAP. Paraesthesia or pain in the distal portion of the upper or lower extremities was the initial complaint in 37 (74%) patients. During disease progression, painful sensations became evident in many of the patients. Indeed, a total of 34 (68%) patients reported pain during the follow-up period. Finally, pain in the extremities became severe and significantly disturbed the quality of life in nine patients. Pacemaker implantation due to complete atrioventricular conduction block was performed prior to the diagnosis of FAP ATTR Val30Met in three (6%) patients, and post-diagnosis in another three (6%) patients. The age of pacemaker implantation was 68.7±2.8 years. Ocular amyloidosis was observed in five (10%) patients, while hoarseness of the voice was reported by 11 (22%) patients, among whom this symptom first appeared at 68.0±5.8 years of age.
Backgrounds and clinical characteristics
Appearance of clinical landmarks according to age
The distribution of ages at which each of the clinical landmarks initially appeared is summarised in figure 1. Overall, the age of onset of each clinical landmark varied by a wide margin of approximately 12 years when assessed by the mean±SD range. However, the mean ages of onset of a given clinical landmark were distributed within a narrow range of approximately 5 years. In addition, the order of appearance of each clinical landmark tended to be uniform between patients. The average age at which sensory and motor symptoms in the upper and lower extremities appeared tended to be concentrated in a relatively narrow age window of less than one and a half years. Generally, lower limb sensory symptoms were the earliest of the clinical landmarks, occurring at 65.1±6.5 years of age. On the other hand, upper limb sensory symptoms appeared at 66.0±6.1 years of age. Among five (10%) of patients did upper limb sensory symptoms precede lower limb sensory symptoms and present as the initial symptom of the disease. Interestingly, the appearance of upper limb motor symptoms occurred at 66.3±5.8 years of age, thereby slightly preceding that of lower limb motor symptoms, which occurred at 66.5±6.2 years of age. Digestive and orthostatic symptoms tended to appear during the early period of disease (66.4±5.8 and 66.4±6.3 years of age, respectively). Orthostatic symptoms became exacerbated further on during follow-up, resulting in significant disturbance in the activities of daily living. For instance, at this advanced disease stage, some patients experienced syncope even while in a seated position. Urinary symptoms tended to appear later on in the disease process, occurring at 67.4±6.7 years of age. Patients began using a cane and wheelchair at 67.7±6.6 and 68.9±6.0 years of age, respectively. Cardiac symptoms or signs tended to be noted only in the late phase of the disease, at 69.3±6.2 years of age. Based on the data from 21 deceased patients, the age at death was 70.0±6.0 years. No significant differences were observed between male and female patients in the age at which various clinical landmarks appeared. The duration from disease onset to death was 7.3±2.9 years, which ranged widely from 2 to 15 years (table 2). The predominant cause of death was heart failure due to cardiac amyloidosis, accounting for eight of 21 observed deaths. Sudden death occurring mostly outside a hospital was reported in seven patients. Other four patients died of cachexia and secondary infection such as pneumonia and urinary tract infection. The duration from disease onset to death was similar regardless of the cause of mortality.

Appearance of clinical landmarks according to age. Overall, the age of onset of each clinical landmark varied by a wide margin of approximately 12 years when assessed by the mean±SD range. However, the mean ages of onset of a given clinical landmark were distributed within a narrow range of approximately 5 years. Finally, the order of appearance of each clinical landmark tended to be uniform between patients. UL, upper limbs; LL, lower limbs.
The age and cause of death
Laboratory findings
Patient laboratory findings at the time of diagnosis are summarised in table 3. The diagnosis of disease occurred 2.8±2.2 years following the onset of disease. Although anaemia and renal insufficiency were reported in conventional FAP ATTR Val30Met cases from endemic foci,36 a reduction in haemoglobin level and an increase of serum creatinine level were not conspicuous in the examined patients. One of 39 (3%) examined patients had a haemoglobin level of <10 g/dl, while four of 37 (11%) examined patients presented with a creatinine level exceeding 1 mg/dl. Anaemia and renal insufficiency did not become major problems in any patients during the follow-up. Protein levels in cerebrospinal fluid ranged from 21 to 148 mg/dl and exceeded 45 mg/dl without an increase in cell count among 22 of the 41 (54%) examined patients. The findings of nerve conduction studies were mainly indicative of axonal neuropathy predominantly in the lower limbs along with some slowing of conduction velocities and prolongation of distal latency as described previously.17 18 28 Reduction of CMAP and SNAP in the lower limbs was already prominent at the time of diagnosis; CMAPs were not elicited in 19 of 43 (44%) patients in the tibial nerve, while SNAPs were not elicited in 38 of 46 (83%) examined patients in the sural nerve. Cardiomegaly, defined as cardiothoracic ratio exceeding 50% on chest x-ray, was observed in 19 of 30 (63%) examined patients. On echocardiography, a thickened interventricular septum was seen in 21 of 29 (72%) examined patients (normal value, 11 mm or less18). Granular sparkling echo was found in 12 of 24 (50%) examined patients. Levels of serum NT-proBNP were elevated in 16 of 19 (84%) examined patients. However, at the time of diagnosis, symptoms or signs of heart failure were reported in only seven (14%) cases. First or second degrees of atrioventricular conduction block were detected at the time of diagnosis in five of 41 (13%) examined patients.
Laboratory findings at diagnosis
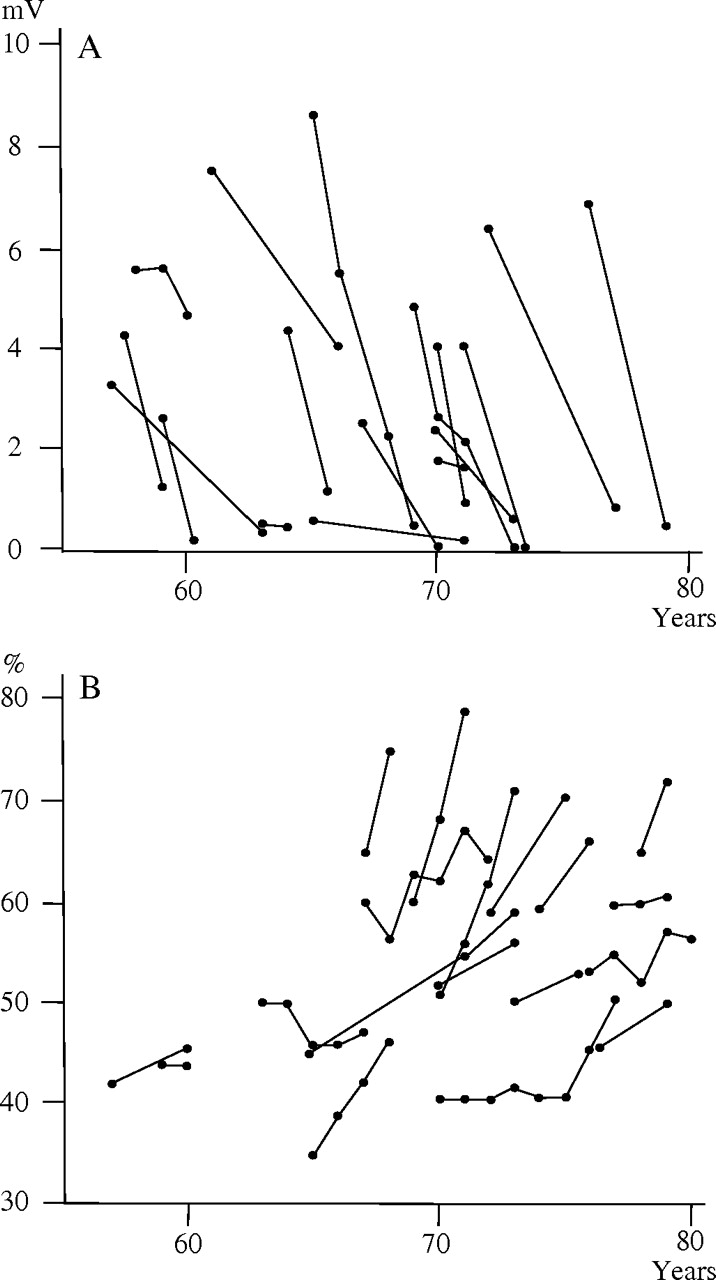
The electrophysiological indices of the median, ulnar, tibial and sural nerves tended to show weak or no correlations with the age at examination or with the time from disease onset to the examination. Indeed, with the exception of an association between MCV in the ulnar nerve and the time from disease onset to the examination (r=−0.602, p<0.0001), Pearson's correlation coefficients for all these associations failed to exceed 0.40. However, serial measurement of electrophysiological indices clearly demonstrated an exacerbation of these indices occurring within a span of a couple of years. For example, serial measurement of CMAP in the median nerve obtained from 17 patients decreased from 3.9±2.1 mV at the first measurement to 1.2±1.3 mV at the subsequent measurement performed 2.7±1.8 years later (figure 2A). At the same time period, MCV and distal latency in the median nerve were aggravated from 48.1±4.9 m/s and 4.4±0.7 ms to 33.8±10.0 m/s and 7.0±2.2 ms, respectively.
{kind=link}
{kind=link}
Serial measurement of compound muscle action potential (CMAP) in the median nerve and cardiothoracic ratio. (A) Serial CMAP obtained from 17 patients indicated steady worsening in individual patients within a span of a couple of years. Specifically, CMAP decreased from 3.9±2.1 mV at the first measurement to 1.2±1.3 mV at the second measurement performed 2.7±1.8 years later. (B) Serial cardiothoracic ratio obtained from 19 patients increased in unison, with the duration of the disease increase from 52.3±8.4% at the first assessment to 59.0±11.0% at the last assessment performed 3.1±1.9 years later.
Similar characteristics were seen in the cardiac indices assessed. The cardiothoracic ratio, the interventricular septum thickness and the serum levels of NT-proBNP showed no correlation with the age at examination or the time between disease onset and examination. On the other hand, serial measurements of the cardiothoracic ratio obtained from 19 patients indicated that the ratio tended to increase in unison with the length of the disease. For instance, the cardiothoracic ratio was 52.3±8.4% at the first assessment and 59.0±11.0% at the last assessment performed 3.1±1.9 years later (figure 2B). Serial values of interventricular septum thickness obtained from 20 patients also indicated an increase from 14.4±4.2 mm at the first assessment to 17.1±5.6 mm at the last assessment performed 3.2±1.5 years later.
Discussion
Our study elucidated the natural history of late-onset FAP ATTR Val30Met among patients from non-endemic areas of Japan. We accomplished this task by focussing on the age of the appearance of major clinical landmarks, as well as longitudinal assessments of electrophysiological and cardiac indices. The importance of the current endeavour is highlighted by the focus on the most common and ubiquitously prevalent form of FAP. Since liver transplantation has become established as a treatment for FAP,19 establishing the natural history of this disease by studying a large number of cases, especially those with an early disease onset residing in the endemic foci, has become a challenge. Based on data obtained prior to the establishment of liver transplantation, the mean duration from the onset to death among patients with FAP ATTR Val30Met from a Japanese endemic focus was 8.8 years.37 According to a prior report by Andrade,38 the duration of disease among Portuguese patients ranged from 7 to 10 years. However, a subsequent report from Portugal described the mean disease duration to be 10.8 years.39 Additionally, the mean disease duration among Swedish patients was reported to be 10.7 years.1 However, subsequent development of treatments for various FAP symptoms may further extend the disease duration.36 To date, the progression of FAP from diagnosis to death is considered to take between 5 and 15 years.26 On the other hand, patients with late-onset FAP ATTR Val30Met from non-endemic areas of Japan seem to progress in a relatively short mean disease duration of 7.3 years (4.3 years from disease diagnosis to death).
The mode of development of FAP ATTR Val30Met clinical symptoms among patients from non-endemic areas has not been previously studied systematically. Although the ages of onset of each clinical landmark were variable, once the initial symptom appeared, the disease was found to progress steadily with a uniform chronological sequence of respective clinical landmarks that occurred over a relatively short time span. The rate of reduction of CMAP in patients with FAP ATTR Val30Met from non-endemic areas also seemed to be faster than that of patients from endemic foci.17 40 Because the mutation of the TTR gene is inherited, a variant TTR is considered to be present from infancy. However, previous studies suggested that the expression level of serum TTR does not influence the age of onset or the severity of the disease.41–43 The clinical and laboratory findings in the present study indicate a rapid deterioration in these indices following disease onset. Which factor determines the age of onset remains to be investigated in future research.
In the present study, once neuropathic symptoms were noted, sensory and motor symptoms in both the upper and lower extremities appeared within a short time period of less than one and a half years. As a result, upper limb symptoms preceded lower limb symptoms in some of our patients. One of them underwent bilateral carpal tunnel release because the patient was diagnosed as carpal tunnel syndrome by an orthopaedist. A previous report also described two older patients from non-endemic areas who underwent carpal tunnel release before diagnosis of FAP ATTR Val30Met.44 The appearance of upper limb symptoms in our patients seems to be earlier than that previously reported in patients with early-onset FAP ATTR Val30Met from endemic foci of Japan and Portugal. Indeed, among these latter cases, lower limb symptoms invariably precede upper limb symptoms, with a chronological separation of several years.39 45 With the exception of a few patients, diarrhoea and/or constipation did not precede symptoms of somatic neuropathy in our series, although these digestive symptoms tended to initiate in the early phase of the disease. This finding is in contrast with observations made among early-onset cases of FAP ATTR Val30Met from endemic areas, where gastrointestinal symptoms were found to constitute the initial symptoms in nearly half of cases.12 39 45 The digestive symptoms seen in patients with FAP ATTR Val30Met from non-endemic areas were mainly those of lower gastrointestinal tract such as diarrhoea and/or constipation in the present study. Upper gastrointestinal tract symptoms such as nausea and vomiting characteristic in patients from endemic foci were not conspicuous.2 36 Only two patients reported anorexia and nausea in the early phase of the disease.
The predominant causes of death among patients with early-onset FAP ATTR Val30Met from endemic foci in Japan and Portugal include cachexia or secondary infection.37–39 Studies of Swedish patients drew attention to the influence of digestive symptoms at the early phase of the disease on shortening the survival time.1 46 In the present study, the most common cause of death was heart failure due to cardiomyopathy induced by amyloid deposition.25 47 On the other hand, cachexia due to malabsorption did not constitute a major cause of death. Although symptoms or signs suggestive of cardiac involvement tended to appear in the later phase of the disease, they were not apparent in most patients at the time of diagnosis. However, according to assessments conducted at diagnosis using chest x-rays, echocardiography and serum NT-proBNP levels, most patients had findings suggestive of cardiac involvement. These findings indicate that subclinical cardiac amyloidosis occurred in most patients from the early phase of the disease. A majority of patients with late-onset FAP ATTR Val30Met from non-endemic areas have no apparent family history of the disease and also tend to present with non-specific neuropathic features. Not surprisingly, clinicians may misdiagnose such patients with chronic inflammatory demyelinating polyneuropathy or neuropathy of undetermined aetiology, thereby leading to an underestimation of FAP ATTR Val30Met.18 Therefore, proper initial evaluation of patients with neuropathy may help to diagnose late-onset FAP ATTR Val30Met, and enable treatment in the early phase of the disease. Cardiac scintigraphy would also be valuable for an early diagnosis of the disease.48
Secondary to heart failure, sudden death, which occurred mostly at home, accounted for a significant number of deceased patients in the present study. Atrioventricular conduction block requiring pacemaker implantation is a common problem among conventional FAP ATTR Val30Met cases from endemic foci.12 36 However, only a small minority of our patients had undergone a pacemaker implantation. This observation may be attributed to the fact that ECG did not reveal atrioventricular conduction block in most patients at the time of diagnosis. Nevertheless, atrioventricular conduction block may be a cause of death in these patients. Alternatively, since syncope due to orthostatic hypotension tended to become a major problem during the late phase of the disease among the patients studied, this complication may have also contributed to sudden death. Therefore, serial examination and treatment of cardiovascular problems among late-onset FAP ATTR Val30Met cases from non-endemic areas are needed to clarify these issues.
Another important issue uncovered in the present study is that pain in the extremities was highly prevalent. The pain in the extremities became severe and significantly disturbed the quality of life among some of our patients. Symptomatic treatment of the pain common among patients with FAP ATTR Val30Met should be exploited.49
The main advantage of the current study is the focus on obvious and easily recognised clinical landmarks. The clinical landmarks that we adopted for this study were selected with the assumption that they could be accurately assessed by us, the patients and the patients' families in a retrospective fashion. They covered major clinical manifestations in late-onset FAP ATTR Val30Met in non-endemic areas, and their concordance between different evaluators' assessments were verified. Nevertheless, the current study also has a limitation that warrants discussion. Since the study was retrospective in design, disease progression was not prospectively assessed in individual patients. Using disease-specific scales able to quantify the clinical involvement and the severity of the disease is recommended to evaluate the progression of the disease and the response to therapy.32 50 Serial assessments of asymptomatic carriers are also needed because they would be valuable for early diagnosis of the disease and determining the indication of therapy for asymptomatic carriers. A prospective study that assesses the progression of FAP ATTR Val30Met is needed to validate our findings of the natural history of the disease.
Acknowledgments
The authors thank Drs T Shimizu, MD (Department of Neurology, Seirei Hamamatsu General Hospital, Hospital, Shizuoka); A Iwasaki, MD (Department of Neurology, Fukaya Red Cross Hospital, Saitama); R Yuasa, MD (Department of Neurology, Sakai Municipal Hospital, Osaka); R Yamada, MD and K Ichikawa, MD (Department of Neurology, Hyogo Prefectural Amagasaki Hospital, Hyogo) for the provision of clinical data.
References
Footnotes
See Editorial commentary p 121
Linked article 301582.
Funding This work was supported by grants from the Ministry of Health, Labour and Welfare (Research on intractable diseases, H23-012) and the Ministry of Education, Culture, Sports, Science and Technology of Japan (Grants-in-Aid for Scientific Research, 21591076).
Competing interests None.
Ethics approval The study was approved by the ethics committee of Nagoya University Graduate School of Medicine.
Provenance and peer review Not commissioned; externally peer reviewed.