Article Text

Abstract
Roughly two thirds of resuscitated cardiac arrests in children and youth are due to inherited heart diseases. The most commonly implicated are the cardiac ion channelopathies long QT syndrome, CPVT (catecholaminergic polymorphic ventricular tachycardia) and Brugada syndrome. Diagnosis is pivotal to further management of the child if he/she survives, and also to other family members who may be at risk. Thorough investigation of the cardiac arrest survivor is essential to either identify or exclude inherited heart disease. If standard cardiac investigation does not reveal a diagnosis, pharmacological provocation tests are needed to unmask electrocardiographic signs of disease, even if, due to severe brain injury, it is planned ultimately to allow a natural death. Examples are the ajmaline/flecainide challenge for Brugada syndrome and epinephrine for CPVT. A supportive, informative and sympathetic approach to the family is essential. An arrhythmia specialist and a cardiac genetic service should be involved early, with storage of DNA and cardiac/genetic investigation of the family. This review proposes a diagnostic algorithm-based approach to the investigation of this increasingly common clinical scenario.
- Cardiology
- Intensive Care
- Genetics
Statistics from Altmetric.com
Introduction
With increased availability of advisory defibrillators, and general community awareness of, and training in, cardiopulmonary resuscitation (CPR), paediatric intensive care units now commonly receive children and youth who have been resuscitated from a cardiac arrest. Much has been written about resuscitation and early management, but there is remarkably little regarding an investigative approach to determine the underlying cause. In particular, in the awful situation of brain death, what tests should be done prior to, and after, allowing a natural death? This review proposes a practical approach to guide clinicians faced with a child who has experienced a cardiac arrest and has been resuscitated. When the young person is brain dead, completion of some pharmacological challenges will take courage and conviction, and much patient explanation to the family and intensive care staff, prior to withdrawal of life support.
How common is sudden natural death in children and youth?
Sudden unexpected natural death (SUD) in 1–20 year olds occurs at a rate of about 33 per million people per year.1 Of these, about half have a known pre-existing condition (most typically epilepsy, cyanotic heart disease and asthma), while half have no known pre-existing conditions.
What conditions cause sudden and unexpected cardiac death in children and youth?
Among those with no known pre-existing conditions, the autopsy fails to reveal a cause in a third, with death presumed to be due to a primary arrhythmic syndrome (see figure 1). This is consistent with 20–35-year-old sudden death victims, one third of whom also have no overt findings at autopsy.2 ,3

Causes of sudden natural death in 1–20 year olds. Data are extracted from a population-based death certificate review of 270 sudden natural deaths from the North East of England.1 (A) 142 cases with a known previous diagnosis. (B) 128 cases with no previous known diagnosis. CVS, cardiovascular system.
The causes which are identified at autopsy are mostly infectious conditions in children and coronary artery disease in young adults. Heart conditions are identified at autopsy in only about 20% of 1–20 year olds, the top three being aortic stenosis, myocarditis and hypertrophic cardiomyopathy (HCM).1
When a pre-existing condition is present, it is imperative to keep an open mind as to whether this was the real cause of death. Among those with a known pre-existing condition (figure 1A), epilepsy in particular should be queried as a final diagnosis. It is well known that primary arrhythmic syndromes such as long QT syndrome, are commonly misdiagnosed as seizure disorders4 ,5 and epilepsy may be ascribed incorrectly as the cause of death in arrhythmic syndromes.6 Infections also might be a trigger for a terminal arrhythmia rather than the true cause of death, particularly in Brugada syndrome.7
Sudden death in infancy (<1 year of age) is not discussed here specifically, but it is established that primary arrhythmic syndromes, particularly long QT syndrome, are the cause in a minority of such deaths (10–15%).8 These may be less likely to have a familial origin; some infants may die early due to de novo mutations too severe to allow life into the reproductive years. However, after a resuscitated cardiac arrest with documented ventricular arrhythmia in infants, the approach should be much the same as in the older child.
Activity at time of death and relationship to cause
A popular misconception is that SUD most commonly occurs with athletic activity. The literature and media interest in young sudden death is heavily biased towards this population. The commonest causes of sudden death among athletes are HCM, arrhythmogenic right ventricular cardiomyopathy (ARVC) and coronary arterial pathology.5
However, undiagnosed HCM causes less than one death per million person years in the population aged 1–20.1 Community-based studies of sudden death in the young reveal that most deaths occur during rest, sleep or usual daily activities rather than during athletic activity.2 ,3 ,9
Overall, 30–50% of SUDs are due to autosomal dominant inherited primary arrhythmic conditions.9–12 These conditions are diagnosed either by genetic testing of autopsy DNA and/or by cardiac evaluation of family members. These conditions are also known as the cardiac ion channelopathies. Long QT syndrome,13 ,14 Brugada syndrome7 and catecholaminergic polymorphic ventricular tachycardia (CPVT)15 are the most common. Death is due to polymorphic ventricular tachycardia (VT) or ventricular fibrillation (VF). Once detected in the living, the risk of death can be greatly reduced with appropriate intervention, meaning that the identification of these familial conditions in the deceased can save the lives of family members.16
Research on survivors of cardiac arrest at a young age
Recent research has shown that about two thirds of survivors of sudden cardiac arrest have inherited heart disease.17 ,18 This diagnostic rate is higher than that following sudden death and was shown to be mainly because the clinician was able to perform cardiac tests on the presenting individual. Most valuable among these tests are the ajmaline challenge for Brugada syndrome (see below) and exercise testing as well as live imaging, particular echocardiography, in those over 14 years of age.
The most comprehensive study, from the Netherlands, compared 69 cardiac arrest survivors (CA) with 140 SUD victims aged 1–50.18 The distribution of the inherited heart conditions discovered was strikingly different. Cardiac ion channelopathies were found in 57% of SUD versus 28% of CA, whereas HCM was found in only 6% of SUD versus 17% of CA. CPVT was found in 17% of SUD and only 2% of CA. One might conclude that a cardiac arrest due to HCM is survived more frequently than one due to an ion channelopathy. Perhaps this is because HCM arrests are usually precipitated by athletic activity and are more likely to be witnessed. However, referral biases (pre and post mortem) might have influence these results.
Age at cardiac arrest was significant in the type of inherited heart disease found. All of the 1–14 year olds who survived a cardiac arrest had a cardiac ion channelopathy.2 In over 14 year olds, ion channelopathies still account for two thirds of cases, the rest being due to a cardiomyopathy or coronary artery disease or anomaly. Of particular note is the strong link between near drowning in a swimmer and either long QT syndrome or CPVT.19
Brain death and cardiac/genetic investigation
When a young person has a cardiac arrest and sustains severe brain damage, the clinical team and family may choose to allow a natural death. There is, understandably, a tendency to step back medically as nature takes its course and the family grieve. However, there is now a medical imperative to ensure that everything is done prior to death to identify the underlying diagnosis, to ensure that other members of the family are screened and then managed appropriately. We must aim to ensure they do not die from the same condition.
It is also imperative to make a firm diagnosis of a non-inherited cause of the arrhythmic death, since this will relieve the family of uncertainty, and the need for expensive and sometimes invasive cardiac tests on multiple family members. Examples are Wolff-Parkinson White (WPW) syndrome, myocarditis or a coronary arterial anomaly.
Initial investigations and summary of approach
Inclusion criteria
Sudden cardiac death (cardiac arrest) generally refers to those with documented unconscious VF or VT who are resuscitated with CPR, usually requiring electrical cardioversion. For practical purposes it includes any child who needed and responded to CPR, unless a respiratory, neurological or other cause is immediately apparent. This is because malignant ventricular arrhythmias can spontaneously revert to sinus rhythm, as happens every time someone with long QT syndrome or CPVT experiences syncope and then recovers. We also need to include near drowning in a strong swimmer, and seizure with documented VT/VF, or any seizure needing CPR unless a concurrent cardiac rhythm strip shows normal sinus rhythm (see box 1 and figure 2).
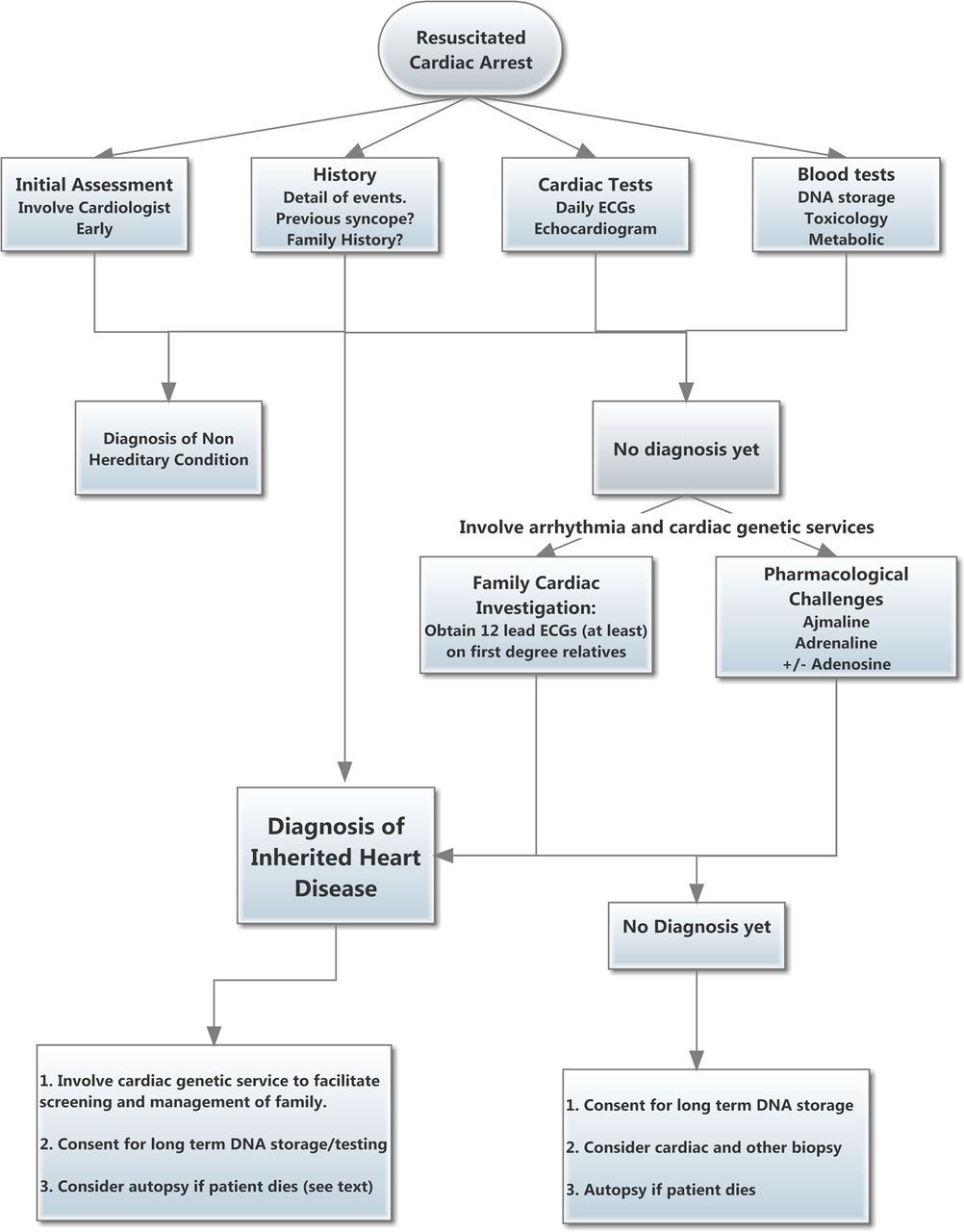
Flow diagram summarising recommended investigative process after resuscitated cardiac arrest. See also box 1.
Infants (under 1 year of age) who should be included are those who had a witnessed sudden collapse without airway obstruction, and clearly any with documented VT or VF. The highest index of suspicion should be for those outside the 1–5-month peak age for sudden infant death, and without other known risk factors for sudden infant death syndrome (SIDS).
Summary of investigative protocol following resuscitated cardiac arrest in children and youth*
-
Inclusion criteria
-
Documented unconscious ventricular fibrillation (VF) or ventricular tachycardia (VT)
-
Any child who needed and responded to cardiopulmonary resuscitation (CPR) (where no neurological, respiratory or other cause is immediately apparent; see text)
-
Near drowning in a strong swimmer
-
Seizure with documented VT/VF or needing CPR
-
Initial assessment
-
A paediatric cardiologist, ideally an electrophysiologist, should be involved early.
-
Review and copy ambulance/defibrillator rhythm strips and get a clear history on the resuscitation from those who gave it.
-
History
-
Document a clear history on presentation-activity at the time, and arrhythmic triggers including recent illness and presence of fever, medications and drugs.
-
Previous history of syncope or seizure?
-
Any previous cardiac tests done?
-
Family history
-
Draw up a family tree asking specifically for history of sudden death in young people, seizures (long QT and other channelopathies are commonly mistaken for epilepsy), syncope, and mention specifically the inherited heart diseases.
-
Cardiac tests
-
Obtain a number of 12-lead ECGs, at least daily initially, document temperature, electrolytes and QT prolonging medication at the time, write these on the ECG (hypothermia, low K+, Mg2+, Ca2+, amiodarone prolongs the QT interval, fever may unmask the Brugada sign). Look in particular for pre-excitation, the Brugada sign, repolarisation abnormalities, ischaemia, and measure the QT interval in at least V5 and lead II.
-
Echocardiogram: look carefully for coronary arterial anomaly and cardiomyopathy in particular.
-
Consider endomyocardial biopsy if history, echocardiogram and ECG are suspicious for myocarditis. Cardiac MRI can also be helpful.
-
Blood tests
-
DNA extraction and storage (EDTA sample). Save blood for possible toxicology and metabolic screen and virology.
-
Family investigations†
-
Seek input of the local cardiac genetic service. Explain to the family about inherited disease as a possible cause.
-
Obtain expert review of a 12-lead ECG on both parents and all siblings. Further tests may be needed depending on the findings in the presenting child.
-
Provocative tests
-
If no diagnosis is achieved yet, provocative tests are required, especially if it is planned to allow natural death. The family may find this stressful and they will need a clear explanation and guidance. It is wise to obtain the family's formal consent.
-
Epinephrine. Important if event occurred with exercise (especially swimming) or excitement. Epinephrine is given centrally at 0.05 µg/kg/min increasing in 5 min intervals by 0.1 and 0.2 µg/kg/min. 12-Lead ECGs are taken at baseline and at the end of each stage. A QTc over 0.5 s is considered to be diagnostic of long QT syndrome25; CPVT is diagnosed by ventricular ectopy, bidirectional VT or polymorphic VT, expert review is essential.
-
Ajmaline. Especially important in cardiac events which occur at sleep/rest or during fever. Unmasks the Brugada signature in those with the syndrome. The sensitivity of the test is improved by moving leads V4–6 (which are not needed) and placing them one intercostal space above V1-V3; rename them as V1–3 upper. Infuse 1 mg/kg ajmaline over 10 min; stop early if positive to minimise risk of inducing VF.
-
Adenosine. By blocking AV nodal conduction transiently, this will unmask concealed pre-excitation due to an accessory pathway. Give 300 µg/kg/min by rapid bolus into a central vein. If heart block or pre-excitation is not seen, give a larger dose until it is.
*Acute life threatening events in infancy rarely fall into this category, where airway, respiratory and neurological issues predominate (see text).
†Family investigations may not be necessary if a non-heritable diagnosis has been made. However, often there is doubt for a considerable time, particularly during cerebral cooling, or after amiodarone when the QT interval may be very long. Furthermore, it is likely that many family members will be present and available for testing; it can be difficult to achieve this later.
Exclusions
Excluded from sudden cardiac death are asystole and extreme bradycardia, which are common terminal events in infancy, where the primary cause is typically neurological/developmental or airway related. Also excluded are most infants who have experienced an acute life threatening event responding to stimulation or airway manoeuvres, where cardiac causes are rare.
Complete atrioventricular block is rarely a cause of community cardiac arrest, although it can occur with acute myocarditis. Since it is immediately apparent on ECG, it is therefore excluded here.
Investigations
Obtain a detailed clinical history, noting activity at time of death and any triggers. Look for a family history for young sudden death and review and copy ambulance data and rhythm strips.
Obtain the ongoing advice of a paediatric cardiologist. Initiate daily 12-lead ECGs, noting body temperature, concurrent QT prolonging medication and serum electrolytes, and obtain expert review of the ECGs and arrange a thorough echocardiogram. Confident exclusion or diagnosis of anomalous coronary artery may require further imaging (such as high resolution CT). Seek evidence of recent viral illness and obtain blood for toxicology and DNA extraction.
This initial phase may reveal a diagnosis. If it does not, further tests are required. These should include at least 12-lead ECGs on first degree relatives with expert review of the results, and some pharmacological challenges in the presenting child. At this stage an arrhythmia specialist should be involved, and the local cardiac genetic service consulted.
Diagnosis of concealed arrhythmic conditions by pharmacological challenge
Long QT syndrome
A third of carriers of long QT syndrome can have a QT interval within the normal range. Among cardiac arrest survivors with long QT syndrome however, the QT interval is usually overtly prolonged, and the repolarisation may look quite abnormal. Repeating an ECG at different times will increase the chance of obtaining one which is diagnostic, and help clarify which changes are due to myocardial stunning from the cardiac arrest. It is imperative that the patient has no QT prolonging factors such as hypothermia or hypokalaemia at the time; these factors should be recorded on each ECG.
In uncertainty, exercise testing20 ,21 or if this is not possible, epinephrine challenge17 ,22 ,23 can help. After exercise or epinephrine, the heart-rate corrected QT interval prolongs paradoxically in the commonest form, type 1. Type 1 long QT syndrome tends to present with near drowning24 and following exercise, particularly in boys. Epinephrine at low dose also induces typical T wave morphology changes (‘notched’) in type 2 long QT syndrome.
Epinephrine is given centrally at 0.05 µg/kg/min increasing at 5 min intervals by 0.1 and 0.2 µg/kg/min. 12-Lead ECGs are taken at baseline and at the end of each stage. A QTc over 0.50 s is considered to be diagnostic of long QT syndrome25; expert review is essential.
No changes are seen with type 3, which usually causes cardiac arrest during sleep. In this uncommon type, the QT interval prolongs during bradycardia and a Holter can be diagnostic.

Three lead electrocardiogram during exercise, typical of catecholaminergic polymorphic ventricular tachycardia (CPVT). Such findings also occur with epinephrine or isoprenaline infusion. Note frequent ventricular couplets, and a run of ventricular tachycardia (VT) at the end of the strip. The VT demonstrates ‘bidirectionality’, pathognomonic of CPVT, with the QRS axis swinging 180 degrees with alternate beats.
CPVT
Since the resting ECG is normal, stress testing with exercise,26 epinephrine or isoprenaline is essential to diagnose CPVT, a highly malignant condition.15 Ventricular ectopy, bidirectional VT and polymorphic VT may be induced (see figure 3). Exercise or isoprenaline may also induce a narrow complex tachycardia in CPVT, including atrial flutter or atrial fibrillation, although these are usually also associated with some element of VT.27 ,28
Brugada syndrome
The Brugada ECG signature, a right bundle branch block appearance with ST elevation seen in the anterior chest leads, can vary enormously with time. In children with Brugada syndrome, fever can unmask the Brugada signature, and VT and VF are usually triggered by high fever; the resultant hypoxic seizure may lead to the erroneous diagnosis of febrile convulsion.7 ,29
Always try to obtain a 12-lead ECG during spontaneous fever.
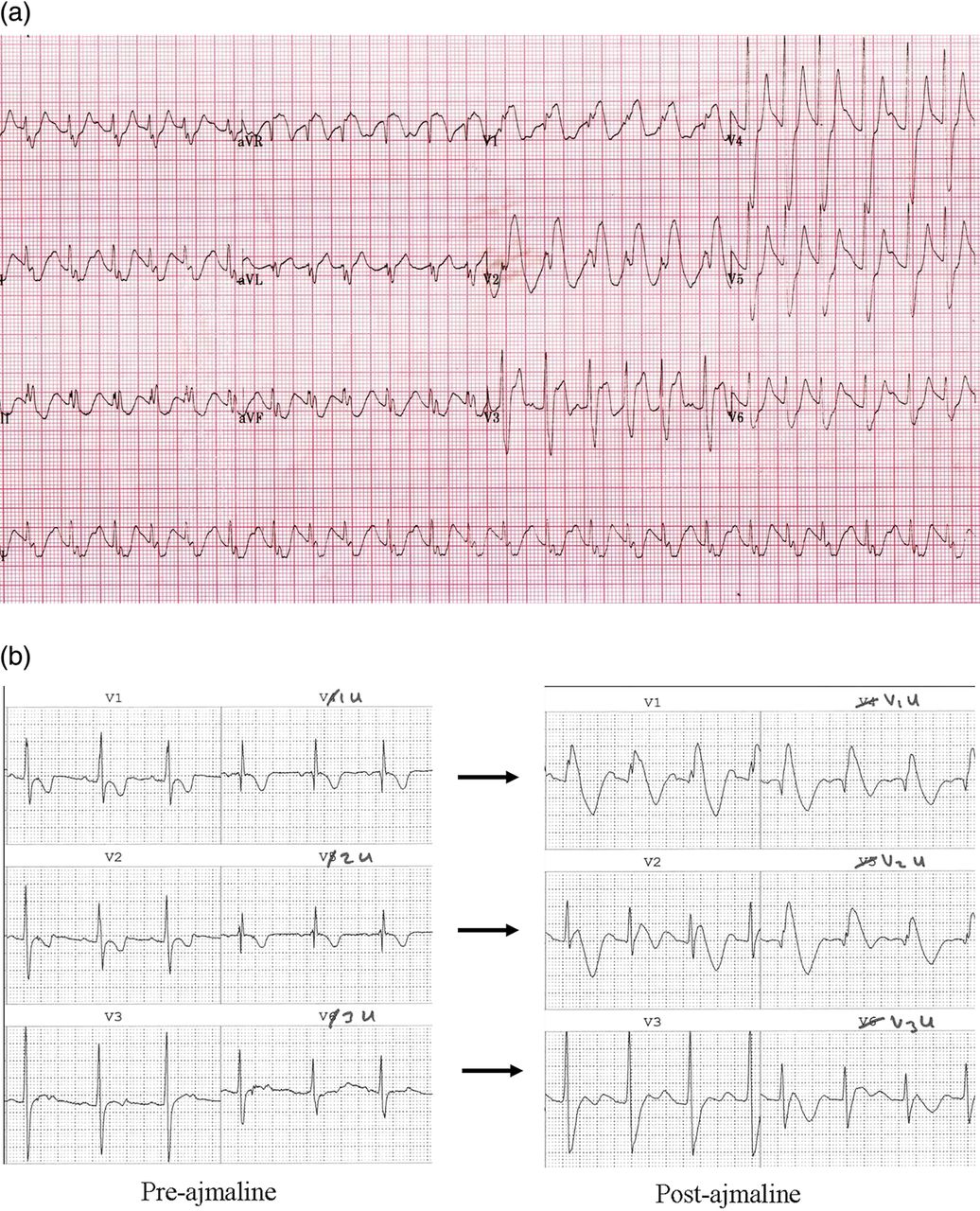
Since the genetic defect relates to decreased functional reserve of the cardiac sodium channel INa, sodium channel blockers can reveal it. Ajmaline (1 mg/kg over 10 min) is preferred to flecainide since it is very short acting and thus safer, and also has better sensitivity.30–32 See figure 4.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Brugada syndrome. (A) This ECG was taken from an 8-month-old infant during fever following cardiac arrest. There is sinus tachycardia with a broad QRS complex of right bundle branch block type, with ST elevation in V1–V3. (B) Positive ajmaline challenge in a 5-year-old child. Chest leads V1–3 are shown, as well as ‘V1u–V3u’. The ‘u’ stands for ‘upper’; chest leads V4–V6 have been removed from their usual position and placed one intercostal space higher than V1–V3. On the left panel there is a partial right bundle branch block pattern. On the right, after ajmaline infusion there is a diagnostic type 1 Brugada sign, best seen in V1, V1u and V2u.
Revealing concealed WPW
WPW is known to cause sudden cardiac arrest when atrial fibrillation is transmitted rapidly to the ventricle.33 In most cases, the delta wave is easily seen in sinus rhythm. In a minority the delta wave is subtle, or even absent. This is commonest when the AV node conducts quickly (such as during inotrope infusion), and the accessory pathway is remote from the AV node, typically leftward and lateral to the mitral valve.
The absence of the normal small Q wave in V5 and V6 should raise suspicion.
An injection of adenosine (300 µg/kg as a rapid central bolus) will block the AV node and manifest the pathway if it is present. If there is no pathway, brief AV block will occur.
Autopsy
In every case, ensure ante mortem DNA is saved with parental consent (5–10 ml of whole blood in an EDTA container). Genetic screening of family members is likely to depend on the diagnosis made from this sample.
If a confident diagnosis of cardiac ion channelopathy has been made, such as long QT syndrome, an autopsy is unlikely to be contributory.
If a cardiomyopathy has been diagnosed, an autopsy may still be helpful depending on the level of certainty, and on the findings in the relatives. An autopsy can reveal multisystem disease which can be otherwise missed and can mimic hypertrophic or dilated cardiomyopathy (eg, metabolic diseases such as Fabry's disease). Also the autopsy may find evidence of myocarditis, and disorders of skeletal muscle. A limited autopsy could be discussed, for example limited to the heart, or one which is minimally invasive with CT or MRI imaging and where tissue biopsies are taken. Second best would be a cardiac and/or skeletal muscle biopsy prior to death.
If the diagnosis is unknown or uncertain, a full autopsy is mandatory.
Genetic testing
The choice of genes for molecular analysis should be guided by a cardiac genetic service which can factor in both the clinical scenario and the costs and practicalities of genetic testing. This is a rapidly evolving field and very soon it will be cost efficient to screen large numbers of genes rather than a select few. However, in 2012, in autopsy negative sudden death, where evaluation of first degree relatives is inconclusive or cannot be completed, current evidence would support analysis of the commoner long QT genes (linked to long QT types 1, 2 and 3, and possibly 5, 6 and 7) and the commonest gene linked to CPVT, RyR2 (the cardiac ryanodine gene).9 ,10 ,34 ,35 The commonest gene linked to familial Brugada syndrome, SCN5A (the cardiac sodium channel gene), is also linked to long QT type 3 and so is already included in this profile.
Conclusion
The tragedy of a young sudden death or of a cardiac arrest with non-survivable brain damage, must not be made worse by clinicians missing the opportunity to identify, or exclude, a lethal familial condition. Every reasonable effort must be made to make a firm diagnosis.
This review arises from part of a series of presentations to the Paediatric Cardiac Intensive Care Society in Cambridge, UK in September 2011.
References
Footnotes
-
Funding Dr Skinner is part funded by Cure Kids.
-
Competing interests None.
-
Provenance and peer review Commissioned; internally peer reviewed.