Article Text

Abstract
Objectives Hydroxychloroquine (HCQ) has been used for decades to treat patients with rheumatic diseases, for example, systemic lupus erythematosus (SLE), rheumatoid arthritis or the antiphospholipid syndrome (APS). We hypothesise that HCQ might target endosomal NADPH oxidase (NOX), which is involved in the signal transduction of cytokines as well as antiphospholipid antibodies (aPL).
Methods For in vitro experiments, monocytic cells were stimulated with tumour necrosis factor α (TNFα), interleukin-1β (IL-1β) or a human monoclonal aPL and the activity of NOX was determined by flow cytometry. The expression of genes known to be induced by these stimuli was quantified by quantitative reverse transcription PCR. Live cell imaging was performed by confocal laser scanning microscopy. Finally, the effects of HCQ on NOX-induced signal transduction were analysed in an in vivo model of venous thrombosis.
Results HCQ strongly reduces or completely prevents the induction of endosomal NOX by TNFα, IL-1β and aPL in human monocytes and MonoMac1 cells. As a consequence, induction of downstream genes by these stimuli is reduced or abrogated. This effect of HCQ is not mediated by direct interference with the agonists but by inhibiting the translocation of the catalytic subunit of NOX2 (gp91phox) into the endosome. In vivo, HCQ protects mice from aPL-induced and NOX2-mediated thrombus formation.
Conclusions We describe here a novel mechanism of action of HCQ, that is, interference with the assembly of endosomal NOX2. Since endosomal NOX2 is involved in many inflammatory and prothrombotic signalling pathways, this activity of HCQ might explain many of its beneficial effects in rheumatic diseases including the APS.
- DMARDs (synthetic)
- TNF-alpha
- Antiphospholipid Antibodies
- Cytokines
- Rheumatoid Arthritis
Statistics from Altmetric.com
Introduction
Antimalarial drugs and in particular chloroquine and hydroxychloroquine (HCQ) have been used for decades in the treatment of rheumatic and autoimmune diseases.1–4 The efficacy of HCQ in patients with systemic lupus erythematosus (SLE) or mild rheumatoid arthritis has been well documented even though the effects are moderate compared with more potent immunosuppressant drugs.5–7 HCQ reduces the risk for thromboembolic events in patients with SLE and positive for antiphospholipid antibodies (aPL).8–10 Accordingly, it has been recommended recently that patients with SLE and aPL should be treated with HCQ as a prophylactic measure.11
While several potential mechanisms of action have been identified, the exact mode of action of HCQ is still under debate.3 ,5 Furthermore, it is not known, if the efficacy of HCQ in different rheumatic and autoimmune diseases is mediated by different mechanisms. The previously described actions of HCQ include reduction of cytokine production,12–14 inhibition of immune effector cells,3 inhibition of platelet function,15 protection of the cell surface from external disturbances,16 competitive binding to nucleic acid ligands of toll-like receptors (TLRs),17 interference with lysosomal function3 ,5 and reduction of leakage of lysosomal enzymes.3 While some of these quite heterogeneous effects associated with HCQ might be related to each other, no unifying mechanism of action of HCQ has been found.
Due to high affinity of HCQ to the lysosomal/endosomal compartment, a potentially relevant target for HCQ might be endosomal NADPH oxidase (NOX). This enzyme complex is involved in numerous proinflammatory signalling cascades.18 In particular, signalling of tumour necrosis factor α (TNFα) via TNF-receptor 1 (TNFR1) and interleukin-1β (IL-1β) via IL-1R are mediated in part by uptake of the ligand-receptor complexes into the endosome, activation of endosomal NOX and generation of superoxide and subsequently other reactive oxygen species (ROS).19–21 Inhibition of endosomal NOX massively reduces downstream activation of NFκB via these pathways. It should be noted though that signalling still proceeds with reduced intensity indicating that the endosomal route accounts for only part of the cytokine effects.18
We have recently shown that aPL also induce endosomal NOX in monocytes and endothelial cells providing a novel mechanism of action of aPL.22–24 Indeed, ability of certain aPL to induce venous thrombosis in vivo depends on NOX2.25 In our hands, aPL induced a more rapid and intense activation of endosomal NOX than TNFα or IL-1β reflected in a much stronger ROS signal. Thus, aPL are another activator of endosomal NOX.
Since inhibition of TNFα and also type 1 interleukins and their signalling pathways has been shown to be therapeutically effective in rheumatoid arthritis2 and probably other autoimmune diseases including SLE26 ,27 and HCQ shows therapeutic benefits in these diseases as well as in antiphospholipid syndrome (APS), we hypothesised that endosomal NOX might be targeted by HCQ. If this hypothesis is correct, it might help to explain many of the observed in vivo effects of HCQ and the efficacy of HCQ in several autoimmune diseases.
Materials and methods
Human aPL
The human monoclonal aPL HL5B has been previously described in detail.28–30 It was cloned from a patient with primary APS and has numerous somatic mutations. It binds to phospholipids in a cofactor-independent manner. An unspecific IgG was generated by the same method. IgG fractions of patients and controls were obtained as described previously (see online supplementary table S1).24 ,31 All antibody and IgG preparations had <0.1 U endotoxin/mL as determined by limulus amoebocyte assay. All patients provided informed consent according to the ethical guidelines following the Declaration of Helsinki. Collection and use of blood samples has been approved by the ethics committee of the State Medical Association of Rheinland-Pfalz.
supplementary figures and table
Cell culture and stimulation of cells
MonoMac1 (MM1) cells were maintained in RPMI-1640 medium supplemented with 10% fetal calf serum, L-glutamine and sodium pyruvate. Monoclonal aPL (100 ng/mL), IgG fractions (100 µg/mL), TNFα or IL-1β (both ebioscience, 10 ng/mL) were added as indicated to 0.5×106 cells/mL. HCQ (Sigma Aldrich, usually 10 µM or as indicated) was added 15 min before the stimuli. To analyse gene expression, RNA was isolated and quantitative reverse transcriptase PCR was performed as previously described.31 Similarly, isolation and culture of human monocytes and mouse monocytes from C57BL/6J mice and gp91phox−/− mice on the same genetic background have been described.23 ,24
Flow cytometric detection of cellular ROS formation
The detection of endosomal ROS generation was performed by use of the fluorogenic reagent OxyBURST Green H2HFF-BSA.32 Cells were kept in phosphate-buffered saline containing 1 mM Ca2+, 1.5 mM Mg2+ and 5.5 mM glucose for 2 hours before being incubated in 10 µg/mL H2HFF-BSA for 2 min with or without HCQ (10 µM) at 37°C. Thereafter, cells were stimulated with agonists as indicated. ROS-induced cellular fluorescence was analysed by flow cytometry.
Confocal laser scanning microscopy
Microscopy was performed with a Zeiss LSM 710 NLO confocal laser scanning microscope and a 1.4 Oil Dic M27 63×plan apochromat objective (Zeiss). To show the intracellular localisation of NOX2 and TLR8 on stimulation with agonists, cells were fixed and stained with fluorescence-labelled anti-TLR8 or anti-gp91phox. Labelled antibodies against calnexin and EEA-1 were used as markers for the endoplasmic reticulum or early endosomes as described by Latz.33 For live cell microscopy, cells were cultured in RPMI without phenol red, stimulated as indicated and imaged directly in chambers maintained at 37°C (Nunc). Monoclonal aPL (HL5B) and control IgG were labelled with fluorescein isothiocyanate (FITC) by the use of a standard FITC Antibody Labeling Kit (Thermo Fisher Scientific).
In vivo thrombosis model and intravital microscopy
The in vivo model of thrombus formation in the inferior vena cava (IVC) used in this study has been previously described in detail.25 ,34 It is based on severe flow reduction in the IVC. Briefly, in anesthetised mice, a median laparotomy was performed and the IVC was exposed. A permanent narrowing ligature was applied exactly below the left renal vein. Human monoclonal aPL HL5B (1 μg) was injected via a jugular catheter 1 hour before flow reduction in the IVC. HCQ (10 μg) was injected intravenously 2 hours before HL5B injection as indicated. Acridine orange was injected (20 µg) intravenously to stain circulating leucocytes in vivo.34 Murine platelets were isolated from whole blood as described34 and labelled with 20 µg/mL rhodamine B. Thrombus formation was observed and quantified by high-speed real-time intravital fluorescence video microscopy (BX51WI; Olympus).
Statistics
All numerical data are shown as mean±SD. Normal distribution was confirmed using Shapiro-Wilk test. Statistical analyses of the data were performed by Student's t-test for normally distributed data and Wilcoxon test for not normally distributed data. p Values <0.05 were considered statistically significant for single testing.
Results
HCQ blocks endosomal NOX activation
Several stimuli including TNFα, IL-1β and certain aPL, for example, the human monoclonal HL5B, induce endosomal NOX2 in monocytic cells followed by downstream effects.22 ,23 ,32 HCQ has high affinity to acidic compartments, that is, lysosomes and endosomes. Therefore, we analysed its influence on endosomal NOX activation in MM1 cells by flow cytometry. As expected, all three stimuli induced significant ROS production with HL5B giving rise to the most rapid ROS production (figure 1A–C). Niflumic acid (NFA), an inhibitor of chloride channel 3 (ClC3), completely blocked cellular ROS production. Since NFA has been shown previously to selectively prevent superoxide generation by endosomal NOX,32 this confirms that ROS induced by TNFα, IL-1β and HL5B is generated by endosomal NOX. HCQ inhibited ROS production in a dose-dependent manner (figure 1D–F). All effects observed in MM1 cells could be reproduced in primary human monocytes (see online supplementary figure S1).

Effect of hydroxychloroquine (HCQ) on superoxide generation. (A–C) MonoMac1 (MM1) cells were loaded with the reactive oxygen species (ROS) sensitive dye OxyBurst before stimulation. Cells were stimulated for up to 60 min with 100 ng/mL HL5B or IgG (A), 10 ng/mL tumour necrosis factor α (TNFα) (B) or 10 ng/mL interleukin-1β (IL-1β) (C) either alone or together with HCQ (10 µM) or niflumic acid (0.1 mM) and absolute fluorescence recorded at the indicated time points. Data are from six independent experiments measured in duplicate. *p<0.01 agonist versus agonist+HCQ. (D–F) Dose–response curves of HCQ effects on ROS production induced by HL5B (D), TNFα (E) or IL-1β (F) in MM1 cells. An almost maximal response is achieved at 10 µM HCQ.
Gene induction in MM1 cells
To analyse whether blockade of endosomal ROS production by HCQ also affects known cellular responses to TNFα, IL-1β and aPL, we determined the effect of HCQ on the induction of genes known to be rapidly and strongly induced by these three agonists. TNFα induces its own secretion via NFκB.35 ,36 IL-1β stimulates IL-8 release in a NFκB-dependent manner.37 We have previously shown that HL5B rapidly induces tissue factor (TF) expression.23 Again the effects of HCQ were compared with those of NFA. Both substances were added 15 min before the respective agonists. As shown in figure 2, NFA and HCQ were equally efficacious in suppressing gene induction by the three agonists. While the effects of IL-1β and aPL on IL-8 and TF were completely blocked at 10 µM HCQ, it appeared that blockade of TNFα was not fully complete. At 3 µM HCQ, inhibition of the agonists was slightly less but still significant. These effects of HCQ could be confirmed on the protein level (data not shown). In addition HCQ blocks aPL-induced translocation of TLR8 to the endosome (see online supplementary figure S2). Our data also confirm that under the cell culture conditions used all effects of the three agonists strongly depend on endosomal NOX.

Effect of hydroxychloroquine (HCQ) on gene induction. MonoMac1 cells were stimulated for up to 6 hours as described in figure 1. Relative expression of tissue factor mRNA (A), tumour necrosis factor α (TNFα) mRNA (B) and interleukin-8 (IL-8) mRNA (C) was normalised to IgG stimulated (A) or unstimulated cells (B+C) and β-actin expression. Data are from six independent experiments (three experiments for 3 µM HCQ and niflumic acid) measured in duplicate. *p<0.05 agonist versus agonist+HCQ.
HCQ does not affect TNFα and IL-1β signalling in NOX2-deficient cells
To assess the contribution of NOX2 inhibition on the overall effect of HCQ, we compared monocytes from gp91phox−/− and wild type C57BL/6J mice. Responses to TNFα and IL-1β were detectable but reduced by 70%–90% in gp91phox−/− mice. Addition of 10 µM HCQ did not have any effect on the residual response in these cells, while it reduced the response in wild type cells to the level of NOX2-deficient cells (figure 3). This suggests that the effect of HCQ on TNFα and IL-1β signalling is more or less exclusively mediated via its effect on endosomal NOX2.

Hydroxychloroquine (HCQ) has no effect on residual tumour necrosis factor α (TNFα) and interleukin-1β (IL-1β) activity in gp91phox-deficient mice. CD115+ monocytes isolated from C57BL/6 wild type mice (A and C) or gp91phox−/− mice (B and D) were stimulated for up to 6 hours with 10 ng/mL of TNFα (A and B) or IL-1β (C and D). Relative expression of TNFα mRNA (A and B) and IL-8 mRNA (C and D) was normalised to unstimulated cells and β-actin expression. Induction of indicated genes was dramatically reduced in NOX2 deficient animals. However, residual cytokine induction was not affected by addition of HCQ. Data are from three independent experiments measured in duplicate. *p<0.01 agonist versus agonist+HCQ.
Mechanism of NOX inhibition
Activation of endosomal NOX by the three agonists depends on the transport of ligand-receptor complexes or aPL into the endosome. HCQ is a lysosomotropic agent that can possibly prevent clathrin-dependent endocytosis.38 ,39 We, therefore, analysed if HCQ has any influence on aPL internalisation. The effect of HCQ on aPL endocytosis was analysed by confocal microscopy using FITC-labelled HL5B. As shown before,22 HL5B is rapidly internalised into the endosomal route as shown by overlap with LysoTracker, a marker for endosomes and lysosomes. There were no discernible effects of HCQ on the pattern of intracellular distribution of HL5B and LysoTracker, providing evidence that HCQ has no effect on internalisation and endosomal accumulation of the monoclonal aPL HL5B (see online supplementary figure S3).
HCQ blocks gp91phox translocation to early endosomes
NOX2 is a membrane-bound enzyme complex of six subunits that converts molecular oxygen to superoxide. In resting cells, the catalytic units gp91phox and p22phox, collectively referred to as flavocytochrome b558, are integrated in membranes, while the other components remain soluble. On stimulation, the cytosolic proteins migrate to the membrane to assemble the active oxidase.40–42 In monocytes and macrophages, the mature flavocytochrome is found primarily in the plasma membrane but also in the endocytic recycling compartment.43
To visualise the localisation of NOX2, MM1 cells were incubated with a fluorescently labelled antibody against gp91phox and a marker for early endosomes (EEA1). In resting cells or cells incubated with control IgG, there was no detectable overlap between gp91phox and EEA1 (figure 4A–C, left panels). Incubation with HL5B, TNFα or IL-1β rapidly induced movement of gp91phox to the endosome as shown by almost complete colocalisation of gp91phox with EEA1. This is the evidence that the three agonists induce assembly of the active multiprotein complex of NOX2 in endosomes. Addition of HCQ to the cell culture completely blocked the movement of gp91phox to the endosome (figure 4). In fact, the distribution of gp91phox after stimulation with HL5B, TNFα or IL-1β in the presence of HCQ was similar to unstimulated cells. In contrast to HCQ, NFA did not prevent movement of gp91phox to the endosome. As previously described, NFA prevents ROS production by blockade of ClC3. Taken together, these data reveal that TNFα, IL-1β and HL5B induce the translocation of gp91phox to early endosomes in monocytic cells, where the active NOX2 enzyme complex is assembled. This early step in endosomal NOX2 activation is blocked by HCQ.

Hydroxychloroquine (HCQ) blocks gp91phox translocation into the endosome. MonoMac1 cells were stimulated for 30 min with (A) HL5B or control IgG (100 ng/mL), (B) tumour necrosis factor α (TNFα) (10 ng/mL) or (C) interleukin-1β (IL-1β) (10 ng/mL). HCQ (10 µM) or niflumic acid (NFA) (0.1 mM) were added as indicated. After fixation, cells were stained with anti-(α)-gp91phox (green), anti-(α)-EEA1 (red) and 4,6-diamidino-2-phenylindole (DAPI) (blue) and visualised by confocal laser scanning microscopy. In unstimulated or IgG stimulated cells, gp91phox is detected at the cell membrane. Incubation with HL5B, TNFα or IL-1β leads to translocation of gp91phox to the endosome as shown by colocalisation with EEA1. This translocation was prevented by addition of HCQ but not by addition of NFA.
HCQ prevents signalling induced by IgG fractions from patients with APS
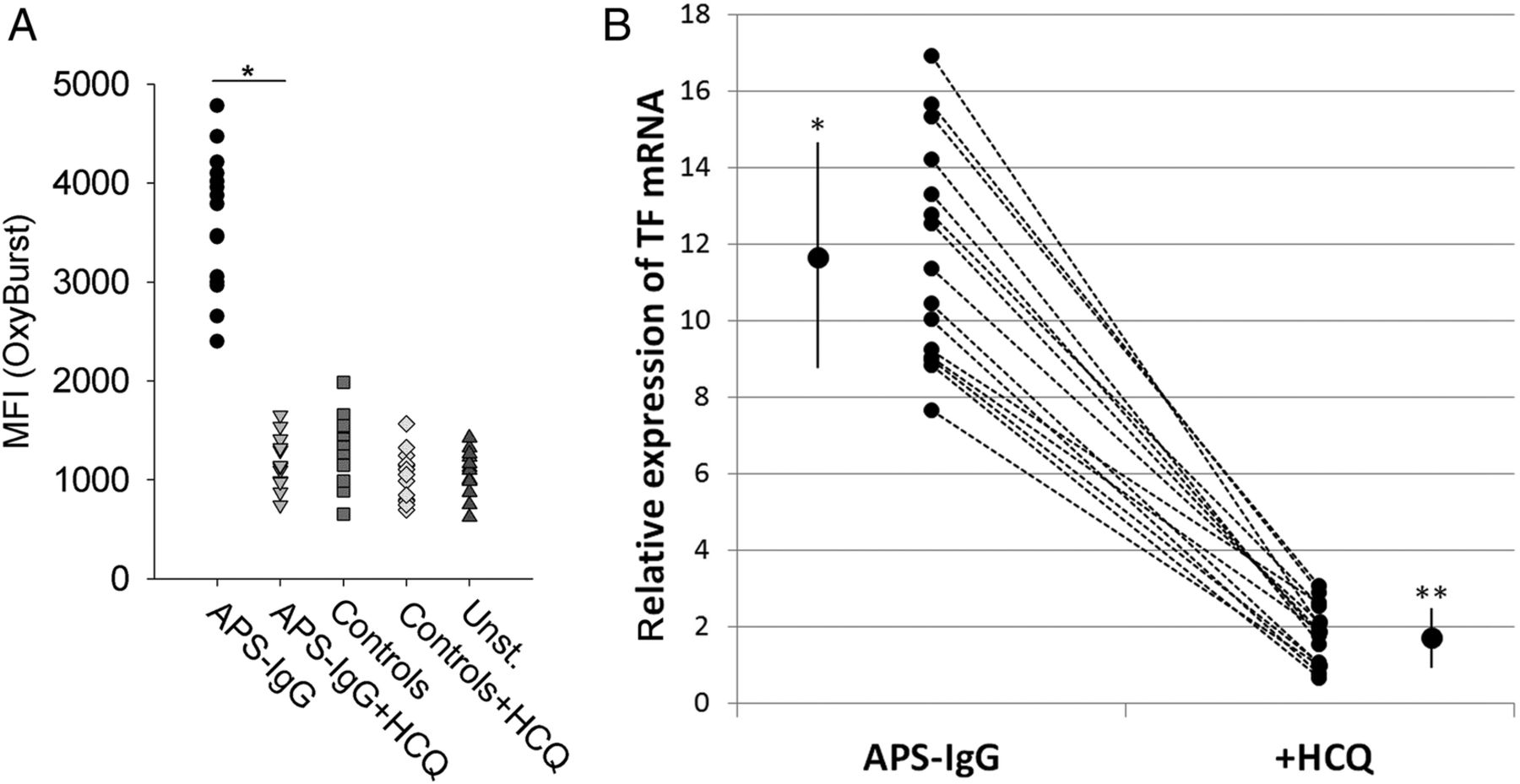
We have previously shown that IgG isolated from 15 patients with APS without exception induced the same signalling cascade as HL5B.24 To exclude that the effect of HCQ was limited to our monoclonal aPL, we confirmed our data using these 15 IgG fractions of patients with APS (see online supplementary table S1). HCQ prevented the increase of endosomal ROS production induced by APS-IgG (figure 5A). As expected, HCQ also almost completely prevented the induction of TF mRNA expression induced by APS-IgG (figure 5B). TF mRNA expression after incubation of MM1 cells with IgG of patients with APS in the presence of HCQ was only slightly higher than mRNA levels of MM1 cells incubated with IgG of 15 healthy control donors (male/female: 5/10; range: 21–72 years).

Hydroxychloroquine (HCQ) prevents reactive oxygen species (ROS) release and tissue factor (TF) induction induced by antiphospholipid syndrome (APS) IgG fractions. MonoMac1 cells were stimulated with IgG fractions (100 µg/mL) of 15 patients with APS and 15 healthy control donors in the absence or presence of HCQ (10 µM) as indicated. (A) Endosomal superoxide production after 20 min of stimulation was detected by flow cytometry using OxyBurst as ROS sensitive dye (*p<0.01). (B) Relative expression of TF (after 1 hour incubation) was normalised to cells stimulated with IgG fractions of 15 healthy control donors (relative mean expression is 1) and to the expression of β-actin. The large symbols indicate mean±SD of the respective normalised mRNA expression. *p<0.0001 versus control IgG and versus HCQ; **p<0.005 versus control IgG.
HCQ prevents HL5B-induced thrombosis in a mouse model
To show that the in vitro effects of HCQ are also relevant in vivo, we chose an in vivo thrombosis model, previously described. This mouse model is based on flow reduction in the IVC and detection of thrombus formation by intravital microscopy. In this mouse model, HL5B and a similar monoclonal aPL, RR7F, massively accelerate venous thrombus formation. This effect is fully dependent on activation of NOX2 as it is absent in gp91phox-deficient mice.25 Pretreatment of the mice with HCQ significantly reduced HL5B-induced venous thrombus formation (figure 6A, B). Together with the in vitro data, this is a strong evidence that inhibition of activation of endosomal NOX by HCQ is also relevant in vivo.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Hydroxychloroquine (HCQ) inhibits thrombus formation induced by HL5B. C57BL/6J mice were infused with HL5B 1 hour before flow reduction in the inferior vena cava. HCQ (10 µg) was infused 2 hours before application of HL5B as indicated. Thrombus formation was visualised by intravital microscopy. (A) Representative microscopic image of the vena cava vessel wall in animals treated with HL5B (upper panel) and HL5B+HCQ (lower panel). Direction of flow is indicated by an arrow. Platelets are labelled red/orange and leucocytes green. (B) Quantitative analysis of thrombus area in HL5B (n=7) and in HL5B+HCQ (n=6) infused mice (*p<0.05).
Discussion
We present a novel mechanism of action how HCQ exerts its therapeutically relevant anti-inflammatory and antithrombotic effects in vitro and in vivo. HCQ blocks a signalling pathway common to TNFα, IL-1β and aPL, which depends on activation of endosomal NOX2 and leads to proinflammatory and procoagulant cellular responses. HCQ concentrations effective in vitro (1–10 µM) are comparable with therapeutic plasma levels of HCQ (3±2 µM). The single dose of HCQ applied in the in vivo mouse model (approx. 0.4 mg/kg body weight) was even lower than therapeutic doses in humans. Interestingly, the major effect of HCQ is the prevention of agonist-induced gp91phox translocation into the endosome. Accordingly, no induction of endosomal NOX2 activity (ie, ROS production) is detectable. This specific extraendosomal effect of HCQ makes a non-specific action by increasing endosomal/lysosomal pH unlikely. As a lysosomotropic weak base, HCQ is rapidly protonated, thereby increasing the pH of endolysosomal vesicles. This may inhibit lysosomal enzymes that require an acidic pH, and prevent fusion of endosomes and lysosomes.44–46 However, a pH optimum for NOX2 between 6.5 and 8.0 has been reported.47 Moreover, it has been shown that NOX2 itself contributes to limiting the acidification of early endosomes in dendritic cells.48 Thus, endosomal NOX2 activity does not depend on acidic pH. Furthermore, it has been shown that 4 µM HCQ affects lysosomal pH minimally17 and in our hands, the labelling pattern of the pH sensitive LysoTracker at 10 µM HCQ is indistinguishable from control.
NOX2 is a membrane-bound enzyme complex generating superoxide. It is made up of the membrane-bound catalytic core (flavocytochrome b558) consisting of the integral proteins p22phox and gp91phox and four cytosolic subunits: p47phox, p67phox, p40phox and Rac1/2.49–51 On stimulation, the soluble factors and flavocytochrome b558 assemble the active enzyme complex and superoxide is released.52 ,53 Thus, NOX2 activity is mainly controlled at the level of the correct and timely assembly of preformed subunits, which is prevented by HCQ.
This mechanism of NOX inhibition by HCQ is obviously different from NFA, an inhibitor of ClC3.32 It has been proposed that ClC3 provides charge neutralisation of NOX-generated electron flux.32 Alternatively, ClC3 may serve as an ion channel for superoxide itself and provide a means to leave the endosome. Thus, NFA does not interfere with NOX2 assembly but inhibits ROS production by the enzyme complex.
In any case, both HCQ and NFA prevent all cellular events downstream of NOX2. This includes induction of TF, TNFα and IL-8 as well as translocation of TLR8 to the endosome. While the induction of cytokines depends on activation of NFκB by ROS, translocation of TLR8 to the endosome induced by aPL depends on superoxide generation but not on NFκB.22 Thus, endosomal ROS-induced downstream effects are not limited to induction of NFκB and genes regulated by this nuclear factor.
To translate our in vitro data to the in vivo situation, we made use of a mouse model of venous thrombosis. We have shown that activation of endosomal NOX2 by aPL greatly accelerates thrombus formation in this model. While wild type C57BL/6J mice rapidly develop venous thrombi when exposed to aPL, gp91phox-deficient mice are protected.25 We show now that HCQ can also prevent aPL-induced thrombus formation in this in vivo model. Thus, our data provide evidence that inhibition of NOX2 activation by HCQ is also a relevant pharmacologic action of this drug in the intact organism.
Inhibition of endosomal NOX2 can explain several well-established effects of HCQ, that is, reduction of cytokine production and plasma concentrations12–14 or inhibition of different immune effector cells.3 Reduced activity of endosomal NOX2 on autocrine or paracrine activation of immune cells by TNFα and IL-1β will lead to reduced production of their target cytokines. Since signalling by TNFα and IL-1β is not exclusively mediated by the endosomal pathway, inhibition by HCQ is most likely incomplete and the effects of HCQ, for example, on signalling by TNFα will be less pronounced compared with effects of direct TNF inhibitors such as adalimumab or etanercept. This is exactly what one might expect from the clinical efficacy profile of HCQ, which is most useful in milder cases of rheumatic diseases. On the other hand, HCQ is able to reduce the effects of other agonists besides TNFα. The relative importance of these pathways with respect to the therapeutic effects of HCQ needs further investigations. This applies particularly to IL-1β, which has been discussed as a potential therapeutic target in rheumatic diseases.
In the case of aPL, we have previously shown that signalling by aPL of similar specificity as our monoclonal aPL HL5B is mediated exclusively via endosomal NOX2. We have elucidated the cellular signalling events induced by HL5B and patient IgG fractions in detail previously.22–25 Their effects are completely blocked by HCQ in vitro and in vivo. In particular, the prevention of thrombus induction in vivo by HCQ is of major relevance. We propose that this effect of HCQ provides an explanation for its beneficial role in the prevention of thromboembolic events in patients with aPL.10 ,11 However, it should be noted that there are other aPL with specificity for β2-glycoprotein I (β2GPI), which have also been shown to be pathogenic in vitro and in vivo.54 These aPL most likely induce cellular responses by other signal transduction pathways, which are probably dependent on formation of a complex of β2GPI/anti-β2GPI. Rand et al have shown that HCQ can disintegrate these complexes and prevent dislocation of annexin A5 from the cell surface. They proposed that this might be an explanation for the protective effect of HCQ.16 Their data imply that the interaction of HCQ with cofactor-independent aPL analysed by us and anti-β2GPI may be quite different. Interestingly, there is some evidence that endosomal uptake of anti-β2GPI may be required for their pathogenic effects.55 However, the relevance of endosomal NOX2 in this process has not yet been analysed.
In conclusion, we present a novel mechanism how HCQ exerts its well-established anti-inflammatory and thromboprophylactic effects. Since signalling endosomes serve as physical platforms for crosstalk between different signalling pathways,56 this might explain the apparently heterogeneous therapeutic profile of HCQ.
Acknowledgments
We thank Dr Steffen Lorenz from the Imaging Core Facility Mainz for providing assistance with confocal microscopy.
References
Footnotes
Handling editor Tore K Kvien
Contributors NM-C designed the study, performed experiments, wrote the manuscript; DM performed in vivo experiments; AC performed experiments; DS performed confocal microscopy; KJL designed the study, wrote the manuscript.
Funding This work was supported by the Federal Ministry of Education and Research (BMBF 01EO1003).
Competing interests None.
Ethics approval Ethikkommission der Landesärztekammer Rheinland-Pfalz.
Provenance and peer review Not commissioned; externally peer reviewed.