





Abstract
Background It is unclear wheather the association between C-reactive protein (CRP) and incident coronary events is free from bias and confounding. Individuals homozygous for a +1444C>T polymorphism in the CRP gene have higher circulating concentrations of CRP. Since the distribution of this polymorphism occurs at random during gamete formation, its association with coronary events should not be biased or confounded.
Methods We calculated the weighted mean difference in CRP between individuals with variants of the +1444C>T polymorphism in the CRP gene among 4659 European men from six studies (genotype-intermediate phenotype studies). We used this difference together with data from previous observational studies to compute an expected odds ratio (OR) for non-fatal myocardial infarction (MI) among individuals homozygous for the T allele. We then performed four new genetic association studies (6201 European men) to obtain a summary OR for the association between the +1444C>T polymorphism and non-fatal MI (genotype-disease studies).
Results CRP was 0.68 mg/l [95% confidence interval (95% CI) 0.31–1.10; P = 0.0001] higher among subjects homozygous for the +1444-T allele, with no confounding by a range of covariates. The expected ORs among TT subjects for non-fatal MI corresponding to this difference in CRP was 1.20 (95% CI 1.07–1.38) using the Reykjavik Heart study data and 1.25 (1.09–1.43) for all observational studies to 2004. The estimate for the observed adjusted-OR for non-fatal MI among TT subjects was 1.01 (95% CI 0.74–1.38), lower than both expected ORs.
Conclusions A common CRP gene polymorphism is associated with important differences in CRP concentrations, free from confounding. The null association of this variant with coronary events suggests possible residual confounding (or reverse causation) in the CRP–coronary event association in observational studies, though the confidence limits are still compatible with a modest causal effect. Additional studies of genotype (or haplotype) and coronary events would help clarify whether or not the link between CRP and coronary events in observational studies is causal.
Prospective studies indicate a robust association, in healthy individuals, between levels of the acute phase reactant C-reactive protein (CRP) and later coronary events,1–3 and it has been proposed that measurement of CRP may be a useful adjunct to coronary risk assessment and that CRP could be causally involved in atheroslerosis.4 However, CRP concentrations are correlated with smoking status, blood pressure, obesity, diabetes, physical activity, social class, low birth weight, and other products of the inflammatory response and are also higher in individuals with clinical cardiovascular disease.5–8 Despite statistical adjustment, this association may, therefore, be subject to residual confounding or bias due to reverse causation, where the disease even in its sub-clinical state leads to elevation in the level of CRP.5–8
A randomized controlled clinical trial of a selective intervention to reduce CRP would provide an unbiased insight into the nature of the association. Unfortunately, no such selective intervention currently exists. Although inhibitors of HMG-CoA reductase (statins) and glitazones reduce CRP,9,10 they also have major effects on lipid profile and glucose metabolism. An alternative approach to control for confounding and reverse causality is to identify a common polymorphism in the CRP gene reliably associated with differences in circulating CRP concentration. The inheritance of such a variant should be subject to the random assortment of maternal and paternal alleles at the time of gamete formation, according to Mendel's second law.11 If CRP actually increases the risk of coronary events then carriage of an allele that exposes individuals to a long-term elevation in CRP should confer an increased risk of coronary events proportional to the difference in CRP attributable to the allele. This relationship should be largely unconfounded and free of reverse causality bias.12,13 Therefore, if non-genetic observational studies were unbiased, the increase in risk estimated from these studies should be consistent with the increase in risk conferred by carriage of the allele.12,13 This approach, known as ‘Mendelian randomization’, has been used recently to investigate the link between homocysteine, fibrinogen, and cardiovascular disease.14–17
CRP concentration is a heritable trait.18,19 In three small studies we found that homozygosity for the rarer T allele of a +1444C>T polymorphism in the 3′-untranslated region of the CRP gene was associated with higher basal and/or stimulated CRP concentrations.20,21 We have now genotyped a large number of Caucasian men to test whether the increase in risk of coronary events in individuals homozygous for this polymorphism is close to that predicted under the assumption that the CRP–coronary event association from previous non-genetic observational studies is free from residual confounding and bias due to reverse causation.
Methods
Study populations
After obtaining ethical approvals, genotyping for the CRP polymorphism and measurement of plasma CRP concentration were conducted in samples obtained from a number of cross-sectional or prospective studies, or randomized controlled trials summarized in Table 1 and in supplementary materials available online.21–27 To test the association of +1444C>T genotype with CRP concentration, male subjects without clinically evident cardiovascular disease from six studies with available DNA and plasma samples were studied (Table 1).21–23,25–27 A second analysis was conducted to evaluate the association between genotype and risk of myocardial infarction (MI) in male subjects from four studies (Table 1).22–25
Design characteristics of the original studies included in the present analysis
| Primary analysis | ||||||||
|---|---|---|---|---|---|---|---|---|
| Name of the study | Study design and median follow-up (years) | Sample size Original/Genotyped | Country(ies) | Study populationa | Main exclusion criteria | Genotype and CRP level | Genotype and non-fatal MI | |
| NPHS-II22 | Prospective cohort (10.6) | 3012/2676 | UKb | Healthy middle-aged men | Pre-existing cardiovascular disease | Yes (2221) | Yes (2676) | |
| Coronary surgery | ||||||||
| Aspirin or anticoagulant therapy | ||||||||
| Malignant disease | ||||||||
| LEADER24 | Nested case–control study from a clinical trial of bezafibrate treatment (4.6) | 1568/1066 | UK | Men with lower extremity arterial disease | Unstable anginaTotal cholesterol <3.5 or >8.0 mmol/lSignificant renal or hepatic disease or malignant disease | No | Yes (1066) | |
| WOSCOPS23 | Nested case–control in a clinical trial of pravastatin treatment (4.9) | 6595/1451 | UK | Moderately hypercholesterolaemic men | MI, or angina pectoris requiring hospitalizationLife-threatening non-cardiac illness | Yes (1334) | Yes (1451) | |
| HIFMECH25 | Case–control (N/A) | Cases: 533/491 | UK | Cases: male MI survivors | Familial hypercholesterolaemia | Yes (465 controls) | Yes (1008) | |
| Controls: 575/517 | SwedenFranceItaly | Controls: men matched by age and regional areas | Insulin-dependent diabetes mellitus | |||||
| Army21 | Cross-sectional (N/A) | 250/219 | UK | Healthy UK-Army recruits | N/A | Yes (219) | No | |
| UDACS27 | Case–control (N/A) | Controls: 449/348 | UK | Cases: men and women with DMc and cardiovascular disease | N/A | Yes (348 Male controls) | No | |
| Controls: men and women with DM but without cardiovascular disease | ||||||||
| EBCT26 | Case–control (N/A) | Controls: 94/72 | UK | Cases: men and women with type-1 DM | N/A | Yes (72 Male controls) | No | |
| Controls: healthy men and women matched by age and gender | ||||||||
| Primary analysis | ||||||||
|---|---|---|---|---|---|---|---|---|
| Name of the study | Study design and median follow-up (years) | Sample size Original/Genotyped | Country(ies) | Study populationa | Main exclusion criteria | Genotype and CRP level | Genotype and non-fatal MI | |
| NPHS-II22 | Prospective cohort (10.6) | 3012/2676 | UKb | Healthy middle-aged men | Pre-existing cardiovascular disease | Yes (2221) | Yes (2676) | |
| Coronary surgery | ||||||||
| Aspirin or anticoagulant therapy | ||||||||
| Malignant disease | ||||||||
| LEADER24 | Nested case–control study from a clinical trial of bezafibrate treatment (4.6) | 1568/1066 | UK | Men with lower extremity arterial disease | Unstable anginaTotal cholesterol <3.5 or >8.0 mmol/lSignificant renal or hepatic disease or malignant disease | No | Yes (1066) | |
| WOSCOPS23 | Nested case–control in a clinical trial of pravastatin treatment (4.9) | 6595/1451 | UK | Moderately hypercholesterolaemic men | MI, or angina pectoris requiring hospitalizationLife-threatening non-cardiac illness | Yes (1334) | Yes (1451) | |
| HIFMECH25 | Case–control (N/A) | Cases: 533/491 | UK | Cases: male MI survivors | Familial hypercholesterolaemia | Yes (465 controls) | Yes (1008) | |
| Controls: 575/517 | SwedenFranceItaly | Controls: men matched by age and regional areas | Insulin-dependent diabetes mellitus | |||||
| Army21 | Cross-sectional (N/A) | 250/219 | UK | Healthy UK-Army recruits | N/A | Yes (219) | No | |
| UDACS27 | Case–control (N/A) | Controls: 449/348 | UK | Cases: men and women with DMc and cardiovascular disease | N/A | Yes (348 Male controls) | No | |
| Controls: men and women with DM but without cardiovascular disease | ||||||||
| EBCT26 | Case–control (N/A) | Controls: 94/72 | UK | Cases: men and women with type-1 DM | N/A | Yes (72 Male controls) | No | |
| Controls: healthy men and women matched by age and gender | ||||||||
All subjects evaluated were Caucasians.
United Kingdom.
DM: diabetes mellitus.
Design characteristics of the original studies included in the present analysis
| Primary analysis | ||||||||
|---|---|---|---|---|---|---|---|---|
| Name of the study | Study design and median follow-up (years) | Sample size Original/Genotyped | Country(ies) | Study populationa | Main exclusion criteria | Genotype and CRP level | Genotype and non-fatal MI | |
| NPHS-II22 | Prospective cohort (10.6) | 3012/2676 | UKb | Healthy middle-aged men | Pre-existing cardiovascular disease | Yes (2221) | Yes (2676) | |
| Coronary surgery | ||||||||
| Aspirin or anticoagulant therapy | ||||||||
| Malignant disease | ||||||||
| LEADER24 | Nested case–control study from a clinical trial of bezafibrate treatment (4.6) | 1568/1066 | UK | Men with lower extremity arterial disease | Unstable anginaTotal cholesterol <3.5 or >8.0 mmol/lSignificant renal or hepatic disease or malignant disease | No | Yes (1066) | |
| WOSCOPS23 | Nested case–control in a clinical trial of pravastatin treatment (4.9) | 6595/1451 | UK | Moderately hypercholesterolaemic men | MI, or angina pectoris requiring hospitalizationLife-threatening non-cardiac illness | Yes (1334) | Yes (1451) | |
| HIFMECH25 | Case–control (N/A) | Cases: 533/491 | UK | Cases: male MI survivors | Familial hypercholesterolaemia | Yes (465 controls) | Yes (1008) | |
| Controls: 575/517 | SwedenFranceItaly | Controls: men matched by age and regional areas | Insulin-dependent diabetes mellitus | |||||
| Army21 | Cross-sectional (N/A) | 250/219 | UK | Healthy UK-Army recruits | N/A | Yes (219) | No | |
| UDACS27 | Case–control (N/A) | Controls: 449/348 | UK | Cases: men and women with DMc and cardiovascular disease | N/A | Yes (348 Male controls) | No | |
| Controls: men and women with DM but without cardiovascular disease | ||||||||
| EBCT26 | Case–control (N/A) | Controls: 94/72 | UK | Cases: men and women with type-1 DM | N/A | Yes (72 Male controls) | No | |
| Controls: healthy men and women matched by age and gender | ||||||||
| Primary analysis | ||||||||
|---|---|---|---|---|---|---|---|---|
| Name of the study | Study design and median follow-up (years) | Sample size Original/Genotyped | Country(ies) | Study populationa | Main exclusion criteria | Genotype and CRP level | Genotype and non-fatal MI | |
| NPHS-II22 | Prospective cohort (10.6) | 3012/2676 | UKb | Healthy middle-aged men | Pre-existing cardiovascular disease | Yes (2221) | Yes (2676) | |
| Coronary surgery | ||||||||
| Aspirin or anticoagulant therapy | ||||||||
| Malignant disease | ||||||||
| LEADER24 | Nested case–control study from a clinical trial of bezafibrate treatment (4.6) | 1568/1066 | UK | Men with lower extremity arterial disease | Unstable anginaTotal cholesterol <3.5 or >8.0 mmol/lSignificant renal or hepatic disease or malignant disease | No | Yes (1066) | |
| WOSCOPS23 | Nested case–control in a clinical trial of pravastatin treatment (4.9) | 6595/1451 | UK | Moderately hypercholesterolaemic men | MI, or angina pectoris requiring hospitalizationLife-threatening non-cardiac illness | Yes (1334) | Yes (1451) | |
| HIFMECH25 | Case–control (N/A) | Cases: 533/491 | UK | Cases: male MI survivors | Familial hypercholesterolaemia | Yes (465 controls) | Yes (1008) | |
| Controls: 575/517 | SwedenFranceItaly | Controls: men matched by age and regional areas | Insulin-dependent diabetes mellitus | |||||
| Army21 | Cross-sectional (N/A) | 250/219 | UK | Healthy UK-Army recruits | N/A | Yes (219) | No | |
| UDACS27 | Case–control (N/A) | Controls: 449/348 | UK | Cases: men and women with DMc and cardiovascular disease | N/A | Yes (348 Male controls) | No | |
| Controls: men and women with DM but without cardiovascular disease | ||||||||
| EBCT26 | Case–control (N/A) | Controls: 94/72 | UK | Cases: men and women with type-1 DM | N/A | Yes (72 Male controls) | No | |
| Controls: healthy men and women matched by age and gender | ||||||||
All subjects evaluated were Caucasians.
United Kingdom.
DM: diabetes mellitus.
Data collection
Data on sex, mean age, systolic and diastolic blood pressure, body mass index (BMI), smoking status, glucose, lipid profile, alcohol consumption, fibrinogen, and plasma CRP values were obtained from the original studies. Subjects were classified using unified definitions of hypertension, hypercholesterolaemia, type-2 diabetes mellitus, and obesity from the guidelines on primary prevention of the American Heart Association.28 For the studies relating genotype and coronary events,22–25 non-fatal MI according to the WHO criteria29 was considered the primary outcome, as this end-point had been uniformly used across all studies. Analyses of genotype and CRP were limited to male subjects to preserve consistency with available studies of genotype and coronary events, which were all conducted in men.
Laboratory analyses
All studies included in the present report used high sensitivity assays to measure plasma CRP concentrations. For specific details of the assays used please see supplementary materials provided online.
CRP genotyping and examination of linkage disequilibrium
The CRP/+1444C>T single nucleotide polymorphism (SNP; rs1130864) was genotyped by PCR and RFLP analysis using primer pairs described previously21 and the restriction enzymes SduI or Bsp1286I that cleave the 181 bp PCR product into 23bp and 158bp fragments only in the presence of the common C allele. All DNA analysis was performed by staff unaware of the clinical status of the subjects. A public domain resequencing resource (Seattle SNPs; http://pga.mbt.washington.edu/) was accessed to investigate associations between the rs1130864 and other SNPs in the CRP gene, and to ascertain haplotype structure. MEDLINE was also searched to identify published studies reporting linkage disequilibrium (LD) between CRP SNPs and haplotype structure.
Statistical analysis
Genotype and CRP concentration
To quantify the effect of the CRP gene polymorphism on CRP concentration, we genotyped 4659 men from six studies (Table 1).21–23,25–27 We calculated the within-study mean difference in CRP concentration between individuals homozygous for the T allele and carriers of the C allele and then weighted each mean by the inverse of its variance to obtain an overall weighted mean difference (WMD). In the calculation of the WMD, we limited the analysis to subjects without known coronary or peripheral artery disease at the time of blood sampling in order to avoid the potential for established disease to modify the size of the genotype–CRP association.16 However, an analysis including all subjects was also conducted. Therefore, for prospective studies, baseline CRP data were used from all available subjects without clinically evident atherosclerosis. For case–control studies, genotype–CRP associations were analysed solely using control subjects. Because of its skewed distribution, CRP values were log-transformed before the analysis. For methods used to calculate the absolute geometric-WMD in CRP concentrations by genotype please see supplementary materials available at IJE online. Random and fixed effect models were used for these analyses.30,31
Genotype and non-fatal myocardial infarction
To examine the effect of CRP genotype on risk of non-fatal MI, we genotyped 6201 men from four studies (Table 1).22–25 We obtained the adjusted odds ratios (ORs) and 95% confidence intervals (95% CIs) for subjects homozygous for the T allele compared with carriers of the C allele from each study. In addition, for the two intervention trials23,24 the potential interaction of the genotype–MI association with the active therapy was also evaluated. We then pooled the within-study ORs to obtain a summary adjusted-OR and 95% CI for non-fatal MI, under both fixed and random effect models. Fixed effect summary-ORs were calculated using the inverse variance-weighted method,32 and the DerSimonian and Laird Q test33 was used to evaluate the degree of heterogeneity between studies.
Consistency between the ORs from genetic and from non-genetic observational studies
We estimated the expected OR for non-fatal MI, corresponding to the WMD in CRP between TT subjects and C allele carriers, based on data from prior non-genetic observational studies that examined the CRP–coronary event association. To do this we used information from the most recent meta-analysis of observational studies of CRP and coronary events as well as data from the Reykjavik Heart Study, a very large study in which more extensive control for potential confounders was made. The first estimate, based on an OR of 1.58 (95% CI 1.48–1.68), for the top vs bottom tertiles of the CRP distribution, came from the recent meta-analysis34 of 22 prospective studies with 7068 cases with different degrees of adjustment for traditional cardiovascular risk factors across the studies included and no adjustment for regression dilution bias. The second estimate was based on an OR of 1.45 (95% CI 1.26–1.68) between top and bottom tertiles of the CRP distribution reported in the Reykjavik Heart Study,34 the largest single prospective observational study of CRP, which contributed 2459 cases to the meta-analysis and also undertook a comprehensive adjustment for potential confounders (age, sex, period of recruitment, smoking, systolic blood pressure, total cholesterol level, triglyceride levels, BMI, forced expiratory volume in one second, diabetes, and socioeconomic status). The third estimate from the Reykjavik Heart Study34 was based on an OR of 1.92 (95% CI 1.68–2.18) for top vs bottom tertile of CRP, based on a more limited degree of adjustment (age, sex and period of recruitment). The final estimate also from the Reykjavik study34 was based on the adjusted standard 10 year follow-up risk of 1.84 (95% CI 1.49–2.28) for top vs bottom tertile of CRP.
For these estimates we assumed equivalence between relative risk and OR, that the usual mean difference in CRP between the individuals in top and bottom tertiles was 1.4 mg/l6, and that the CRP–coronary event relationship was log-linear. The expected OR for TT homozygous subjects with reference to C allele carriers was calculated using the formula: expected
Results
Allele and genotype frequencies, and LD with other CRP SNPs
The allele and genotype frequencies of the +1444C>T polymorphism were in Hardy–Weinberg equilibrium for all studies included in the present report, apart from a marginal distortion in the NPHS-II study (please see Table 1 in supplementary materials available at IJE online). The frequencies of the rare allele in disease-free subjects from all studies were very similar (range: 26–33%). Data from the Seattle SNPs resequencing resource (http://pga.gs.washington.edu/data/crp) indicates that four common CRP haplotypes (approximate frequencies 0.3, 0.3, 0.3, and 0.05) account for >90% of chromosomes among subjects of European descent. The +1444T allele lies on one of the three common haplotypes (approximate frequency 0.3), which also contains the T allele of a common tri-allelic polymorphism (−286C>T/A; rs3091244). Prior studies also report a strong LD between minor alleles at +1444 and −286 polymorphisms; reported D′ values in Caucasians being 0.97,36 0.91,37 0.91 (unpublished data) and with an r2-value of 0.97.38 In functional studies in vitro, the −286C>T/A SNP appears to account for increased transcriptional activity in studies using promoter–reporter assays. The unique haplotype containing minor alleles at −286 and +1444 has been associated with higher CRP values in two published studies,36,39 and we have identified concordant population haplotype frequencies and associations with CRP in our own recent studies in the NPHS dataset of ∼2700 men (unpublished data).
CRP/+1444C>T polymorphism and CRP concentrations
Using data from 4659 men from six studies, the weighted mean CRP concentration in C-allele carriers without known cardiovascular disease was 2.01 mg/l (95% CI 1.94–2.07). Under a fixed effect model, the geometric-WMD in CRP concentration between individuals homozygous for the T allele compared with carriers of the C allele was 0.68 mg/l (95% CI 0.31–1.10; P = 0.0001). There was no significant between-study heterogeneity [P-value for heterogeneity (PHet) = 0.47; Figure 1]. When alternative models of the effect of genotype on CRP were evaluated, only the TT vs CC comparison was significant [WMD = 0.78 mg/l (0.41–1.20); P < 0.0001], while heterozygosity (CT vs CC) was not [WMD = 0.06 (−0.24 − 0.44); P = 0.66]. When subjects with coronary or peripheral atherosclerosis at the time of blood sampling were included in this analysis (n = 5658), the WMD between individuals homozygous for the T allele compared with carriers of C was very similar [0.74 mg/l (95% CI 0.37–1.10); P < 0.0001]. The values we have obtained are concordant with those obtained from other studies examining associations of CRP SNPs with CRP concentration, taking into account known LD.36–40
![Mean log-CRP and mean difference in log-CRP (mg/l) by study among subjects without known cardiovascular disease. *, represents data for control subjects; †, represents data for subjects with later non-fatal MI; ‡, represents relative difference was obtained by antilog of the difference in log-CRP values; and **, represents Geometric WMD = [(relative difference in CRP concentration between TT homozygotes and C-allele carriers × mean-CRP concentration in C-allele carriers) − mean-CRP concentration in C-allele carriers]](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/ije/35/4/10.1093_ije_dyl041/2/m_dyl041f1.gif?Expires=1716582100&Signature=KL5pTGi1WH29r-RAnDsKMWm6eAA~KKCSrTLnccioHFfCFllgu4gyKc3y6AoC0FRmJkYpjYsi7cdtvYkNeKaQAmyhBwg3sJzFmLUw2rkTWEGp7701fUA5tRTeJWBl8MAQXnbDo3DkTVD4cV1w0FyI2tsdZrctelLeK~e-b~4orS2q6jo6Z6MAv1y2C0ViK7FJNgHar9Q7sdIPR3SIEyjeNZ0bZzcHguo6JhhlDZhJJfXDx2DoXPCVobFAhW92-KNNqrijafV8X2vPqqQuyjcIKdkYDAgYI1FbJ9wiCn96-HTG6lILFuxaAwmLK8lUImbZ98P6VYxk8YmL9MfhL8evyg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Mean log-CRP and mean difference in log-CRP (mg/l) by study among subjects without known cardiovascular disease. *, represents data for control subjects; †, represents data for subjects with later non-fatal MI; ‡, represents relative difference was obtained by antilog of the difference in log-CRP values; and **, represents Geometric WMD = [(relative difference in CRP concentration between TT homozygotes and C-allele carriers × mean-CRP concentration in C-allele carriers) − mean-CRP concentration in C-allele carriers]
To evaluate potential confounding of the genotype–CRP association, we quantified the association between TT genotype and age, systolic and diastolic blood pressure, BMI, current smoking, glucose, alcohol intake, fibrinogen, triglyceride, total and HDL cholesterol. No significant difference was observed for any of these risk factors, with the exception of a slightly higher BMI among TT subjects (mean difference = 0.34 kg/m2; P = 0.02; Table 2). In view of the multiple comparisons made, however, this significance level is not particularly extreme. Moreover, this apparent association was not significant when subjects with and without coronary heart disease were considered together, and it is likely, but not yet certain, to reflect the play of chance.
Cardiovascular risk factor distribution according to the CRP/+1444C>T polymorphism
| Variable | Subjects (studies) | Weighted mean difference (TT minus C-carriers) (95% CI) | P-value |
|---|---|---|---|
| Age (years) | 6358 (7) | 0.17 (−0.19 to 0.52) | 0.35 |
| Systolic blood pressure (mm Hg) | 6356 (7) | −0.31 (−1.78 to 1.16) | 0.68 |
| Diastolic blood pressure (mm Hg) | 6356 (7) | −0.70 (−1.56 to 0.16) | 0.11 |
| Body mass index (kg/m2) | 6359 (7) | 0.34 (0.06–0.62) | 0.02 |
| Total cholesterol (mmol/l) | 6115 (6) | −0.01 (−0.08 to 0.06) | 0.87 |
| HDL-cholesterol (mmol/l) | 4714 (5) | −0.01 (−0.04 to 0.01) | 0.32 |
| Triglyceride (mmol/l) | 6041 (5) | −0.03 (−0.11 to 0.06) | 0.51 |
| Fibrinogen (g/l) | 5656 (4) | 0.03 (−0.02 to 0.08) | 0.20 |
| Glucose (mmol/l) | 1913 (3) | −0.01 (−0.13 to 0.12) | 0.90 |
| Alcohol intake (U/week) | 4549 (4) | 0.01 (−1.38 to 1.39) | 0.99 |
| Current smokinga | 6132 (6) | 1.00 (0.84–1.20) | 0.98 |
| C-reactive protein (mg/l) | 4659 (6) | 0.68 (0.31–1.10) | 0.0001 |
| Variable | Subjects (studies) | Weighted mean difference (TT minus C-carriers) (95% CI) | P-value |
|---|---|---|---|
| Age (years) | 6358 (7) | 0.17 (−0.19 to 0.52) | 0.35 |
| Systolic blood pressure (mm Hg) | 6356 (7) | −0.31 (−1.78 to 1.16) | 0.68 |
| Diastolic blood pressure (mm Hg) | 6356 (7) | −0.70 (−1.56 to 0.16) | 0.11 |
| Body mass index (kg/m2) | 6359 (7) | 0.34 (0.06–0.62) | 0.02 |
| Total cholesterol (mmol/l) | 6115 (6) | −0.01 (−0.08 to 0.06) | 0.87 |
| HDL-cholesterol (mmol/l) | 4714 (5) | −0.01 (−0.04 to 0.01) | 0.32 |
| Triglyceride (mmol/l) | 6041 (5) | −0.03 (−0.11 to 0.06) | 0.51 |
| Fibrinogen (g/l) | 5656 (4) | 0.03 (−0.02 to 0.08) | 0.20 |
| Glucose (mmol/l) | 1913 (3) | −0.01 (−0.13 to 0.12) | 0.90 |
| Alcohol intake (U/week) | 4549 (4) | 0.01 (−1.38 to 1.39) | 0.99 |
| Current smokinga | 6132 (6) | 1.00 (0.84–1.20) | 0.98 |
| C-reactive protein (mg/l) | 4659 (6) | 0.68 (0.31–1.10) | 0.0001 |
Comparisons are made between TT homozygotes and C-allele carriers.
For this variable instead of the WMD, the value reported is the weighted odds ratio.
Cardiovascular risk factor distribution according to the CRP/+1444C>T polymorphism
| Variable | Subjects (studies) | Weighted mean difference (TT minus C-carriers) (95% CI) | P-value |
|---|---|---|---|
| Age (years) | 6358 (7) | 0.17 (−0.19 to 0.52) | 0.35 |
| Systolic blood pressure (mm Hg) | 6356 (7) | −0.31 (−1.78 to 1.16) | 0.68 |
| Diastolic blood pressure (mm Hg) | 6356 (7) | −0.70 (−1.56 to 0.16) | 0.11 |
| Body mass index (kg/m2) | 6359 (7) | 0.34 (0.06–0.62) | 0.02 |
| Total cholesterol (mmol/l) | 6115 (6) | −0.01 (−0.08 to 0.06) | 0.87 |
| HDL-cholesterol (mmol/l) | 4714 (5) | −0.01 (−0.04 to 0.01) | 0.32 |
| Triglyceride (mmol/l) | 6041 (5) | −0.03 (−0.11 to 0.06) | 0.51 |
| Fibrinogen (g/l) | 5656 (4) | 0.03 (−0.02 to 0.08) | 0.20 |
| Glucose (mmol/l) | 1913 (3) | −0.01 (−0.13 to 0.12) | 0.90 |
| Alcohol intake (U/week) | 4549 (4) | 0.01 (−1.38 to 1.39) | 0.99 |
| Current smokinga | 6132 (6) | 1.00 (0.84–1.20) | 0.98 |
| C-reactive protein (mg/l) | 4659 (6) | 0.68 (0.31–1.10) | 0.0001 |
| Variable | Subjects (studies) | Weighted mean difference (TT minus C-carriers) (95% CI) | P-value |
|---|---|---|---|
| Age (years) | 6358 (7) | 0.17 (−0.19 to 0.52) | 0.35 |
| Systolic blood pressure (mm Hg) | 6356 (7) | −0.31 (−1.78 to 1.16) | 0.68 |
| Diastolic blood pressure (mm Hg) | 6356 (7) | −0.70 (−1.56 to 0.16) | 0.11 |
| Body mass index (kg/m2) | 6359 (7) | 0.34 (0.06–0.62) | 0.02 |
| Total cholesterol (mmol/l) | 6115 (6) | −0.01 (−0.08 to 0.06) | 0.87 |
| HDL-cholesterol (mmol/l) | 4714 (5) | −0.01 (−0.04 to 0.01) | 0.32 |
| Triglyceride (mmol/l) | 6041 (5) | −0.03 (−0.11 to 0.06) | 0.51 |
| Fibrinogen (g/l) | 5656 (4) | 0.03 (−0.02 to 0.08) | 0.20 |
| Glucose (mmol/l) | 1913 (3) | −0.01 (−0.13 to 0.12) | 0.90 |
| Alcohol intake (U/week) | 4549 (4) | 0.01 (−1.38 to 1.39) | 0.99 |
| Current smokinga | 6132 (6) | 1.00 (0.84–1.20) | 0.98 |
| C-reactive protein (mg/l) | 4659 (6) | 0.68 (0.31–1.10) | 0.0001 |
Comparisons are made between TT homozygotes and C-allele carriers.
For this variable instead of the WMD, the value reported is the weighted odds ratio.
OR estimated from observational studies and the expected mean difference in plasma CRP
Expected ORs for non-fatal MI among TT homozygotes compared with C allele carriers based on a between-genotype WMD difference in CRP of 0.68 mg/l (95% CI 0.31–1.10) were 1.37 (95% CI 1.14–1.68), 1.20 (95% CI 1.07–1.38), 1.34 (95% CI 1.12–1.67), and 1.25 (95% CI 1.09–1.43) based, respectively, on the Reykjavik Heart Study minimally and maximally adjusted models, the Reykjavik study 10 year estimate, and on the meta-analysis of studies up to 2004.34
OR of non-fatal MI for the CRP/+1444C>T polymorphism
Data from four new studies involving 985 subjects with non-fatal MI and 5216 control subjects were pooled to obtain a summary adjusted OR. After combining the studies under a fixed effect model, subjects homozygous for the T allele compared with C allele carriers had no significant increase in the risk of non-fatal MI [summary adjusted-OR= 1.01 (95% CI 0.74–1.38); P = 0.95] (Figure 2). No significant inter-study heterogeneity was observed (PHet = 0.14). When alalysis of the risk of non-fatal MI conferred by homozygosity for the T allele was restricted to prospective genetic studies, there was still no significant association with non-fatal MI [OR = 0.98 (95% CI 0.70–1.38); P = 0.93]. No significant increase in risk was observed in the individual studies, and there was no significant interaction between treatment and genotype–outcome association in the two clinical trials involved in this analysis: for LEADER, P = 0.53 and WOSCOPS, P = 0.44.
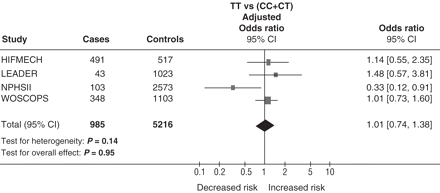
Adjusted odds ratio (a multivariate logistic regression model was used to adjust the OR from each study by age, total cholesterol, alcohol intake, body mass index, hypertension, diabetes, and smoking) for non-fatal MI among subjects with the +1444TT-genotype in comparison with carriers of the +1444C allele
Consistency between the ORs estimated from the mean difference in CRP level and the OR from the genetic study
The estimate of the observed OR from the genetic study [1.01 (95% CI 0.74–1.38)] was mathematically smaller than the expected OR [1.37 (95% CI 1.14–1.68) or 1.34 (95% CI 1.12–1.67)], calculated using data from the minimally adjusted estimate or the 10 year estimate from the Reykjavik Heart Study.34 It was, however, closer to estimates based on data from the maximally adjusted model of the large Reykjavik Heart Study [1.20 (95% CI 1.07–1.38)] and to an estimate based on an updated meta-analysis of studies to 2004 [1.25 (95% CI 1.09–1.43)]. Indeed, most of the 95% confidence limits of these latter two estimates lay within the upper bound of the 95% confidence limit of the genetic estimate (Figure 3). The P-value for interaction for the comparisons of the minimally and maximally adjusted estimates from the Reykjavik study, and from an updated meta-analysis of studies to 2004 with the observed genetic estimate is outlined inFigure 3.
![Comparison of expected and observed odds ratios for non-fatal MI for individuals homozygous for the CRP +1444C>T polymorphism. Expected ORs were calculated assuming a log-linear association between CRP and non-fatal MI, and that a 1.4 mg/l difference in CRP corresponds to an OR for coronary events of 1.92 (95% CI 1.68–2.18) and 1.45 (95% CI 1.25–1.68) based on the minimum and fully adjusted models from the Reykjavik Heart Study published in 2004 and 1.58 (95% CI 1.48–1.68) based on an updated meta-analysis published in 2004, [Data derived from references (6) and (34), respectively]](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/ije/35/4/10.1093_ije_dyl041/2/m_dyl041f3.gif?Expires=1716582100&Signature=OjvGxQJWhBZvTOxev1LUeHANMW6luYbYgrMfiW~Ylphf-hwQoX75AJc3pqAKVaXWIDexCXTTfDr4pWgev6gp5SwRUJW7yVP2iirZ4rjT5XBAFBmJrfE-J6xKQDUP0zli7l5N9lL6ZU1RiNo04qhormXtKW~NSVoit~bl6VJW0LuySqodgi9HQ1QK4JywuOH8cAhc658Eh5J9oewpOwhRIjlge7pXLDJIiarnCAOESMnFxU~FbE5p1XYGwM9H2YRRpA7a-nBKlXTlkQzJ4h7f4Re1~toWDR4cVrnhgaae1dvb3SzNn~CHQkkemqq0UOkimoWTSZlw1WZJ3XHPRW866w__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Comparison of expected and observed odds ratios for non-fatal MI for individuals homozygous for the CRP +1444C>T polymorphism. Expected ORs were calculated assuming a log-linear association between CRP and non-fatal MI, and that a 1.4 mg/l difference in CRP corresponds to an OR for coronary events of 1.92 (95% CI 1.68–2.18) and 1.45 (95% CI 1.25–1.68) based on the minimum and fully adjusted models from the Reykjavik Heart Study published in 2004 and 1.58 (95% CI 1.48–1.68) based on an updated meta-analysis published in 2004, [Data derived from references (6) and (34), respectively]
Discussion
Using pooled data from 4659 individuals, we found that men homozygous for the T allele of the +1444C>T polymorphism of the human CRP gene had a circulating CRP concentration 0.68 mg/l (95% CI 0.31–1.10) higher than carriers of the C allele, confirming the results of earlier smaller studies.20,21 CRP genotype, therefore, contributes to the variation in CRP concentration observed in the population. This association probably reflects LD with a putative functional SNP in the gene promoter (−286C>T/A; rs3091244), although an effect of +1444C>T on RNA stability cannot be ruled out.
We estimated that this difference in CRP of 0.68 mg/l (95% CI 0.31–1.10) would confer an OR for non-fatal MI of 1.25 (95% CI 1.09–1.43) or 1.20 (95% CI 1.07–1.38) using data from a meta-analysis of prospective cohort studies published in 2004,34 and from the maximally adjusted model of the Reykjavik Heart Study,34 respectively, assuming the observational associations have been unbiased and free from residual confounding. However, despite possessing a genotype that would have exposed them to a long-term elevation in CRP concentration, for men with the TT genotype, the point estimate of the OR for non-fatal MI was 1.01 (95% CI 0.74–1.38). This finding is important since, in contrast to the reported association between CRP and coronary events in previous studies, the association between CRP genotype and events should not be subject to reverse causality bias, regression dilution bias, or to confounding by other cardiovascular risk factors related to CRP, though some doubt about a marginal effect of the genotype on BMI remains to be evaluated.7,8,34,41–47 This is supported by the data in Table 2, which indicate that certain established cardiovascular risk factors and some inflammatory markers, that are known to be associated with CRP, were not associated with +1444C>T genotype.
The estimate of risk derived from the genetic association studies of CRP reported here should not be biased or confounded, but it is currently much less precise than the estimates obtained from classical observational epidemiology. It is likely that for this reason an interaction test, which has low power, did not provide formal evidence for heterogeneity between genetic and non-genetic ORs at the 5% level of significance (Figure 3). Approaches for evaluating consistency of genetic and non-genetic disease risk estimates from Mendelian randomization studies have been reviewed recently.48 It is clear, however, from visual inspection of the Forest plot, that the genetic risk estimate is closer to more recent estimates of the CRP–coronary event association reported in an updated meta-analysis of observational studies published in 2004,34 and in the maximally adjusted model of the large Reykjavik Heart Study.34 Thus, recent results from prospective observational studies in which the random error has been reduced substantially by considering more studies in a meta-analysis, and in which more extensive statistical adjustment has been conducted, as exemplified in the Reykjavik Heart Study, indicate that the association of CRP with coronary heart disease risk has decreased substantially in comparison to earlier estimates.34 The strong potential for confounding in observational studies of the measured phenotype (CRP) and the utility of genotype as a proxy for CRP was illustrated by a recent study40 in which a range of covariates that exhibited strong correlations with CRP were distributed evenly among >3500 British women separated according to genotypes for a 1059G>C polymorphism that is in LD with the variant reported here.36
Although a null association was identified between CRP genotype and coronary events in this study the width of the confidence limits is such that our observation could be compatible with a modest but potentially important causal link. It is estimated that a dataset of ∼10 000 cases and similar number of controls would be required in a genetic association study to exclude a small effect of CRP on coronary heart disease aetiology.49 However, evidence from different sources indicates that, if anything, the discrepancy observed between the genetic and non-genetic ORs is likely to be greater, rather than smaller. First, the observational estimate of the CRP–coronary event association34 on which we based the non-genetic OR, did not adjust for regression dilution bias and so may have been underestimated. However, this bias is unlikely to affect the genetic estimate.12 Second, emerging evidence supporting the current null genetic estimate comes from three genetic studies in Caucasians, with an additional 1858 cases and 1347 controls,37,50,51 where carriers of the C allele of the 1059G>C variant (in LD with the +1444/C>T variant) did not have an increased risk of cardiovascular events despite having a CRP concentration that was 0.3–0.6 mg/l higher than GG homozygous subjects. Moreover, recent data from two other genetic studies in Caucasians (703 cases and 1053 controls) found no association of the +1444/C>T variant with cardiovascular events despite a similar effect of this variant on CRP concentration.37,38 An additional approach in future Mendelian randomization studies is to genotype several SNPs at the CRP locus to generate all common haplotypes and to ensure inclusion of SNPs with demonstrated functionality from in vitro studies in such analyses. Since there is extensive LD across the CRP gene, only a small number of ‘tagging’ SNPs (3–5) require typing to capture most of the genetic variation at this locus.52,53 In addition to −286C>T/A, published and public domain data indicate that four common haplotypes identified by these tagging-SNPs account for >90% of chromosomes in subjects of European descent.53
It is important to note that there is evidence for an additional functional CRP promoter SNP found at high frequency in subjects of African origin.36,54
It is important to make clear that our study was limited to male Caucasian subjects and that additional studies will be required to ascertain whether similar findings pertain to women and also to other ethnic groups. However, recent evidence suggests that a similar effect of the +1444C>T polymorphism on CRP concentrations would be expected to occur in women as recently reported in British and American women.38,55 This is also supported by the effect of the 1059/G>C variant on CRP levels, and the 0.42 mg/l difference in CRP concentrations among C carriers compared with women homozygous to the G allele is very similar to the effect observed here in men.38,40
Our finding using a genetic approach also needs to be set in the context of recent mechanistic studies that have investigated a potential pro-atherogenic effect of CRP on vascular cells and tissues.56–61 It is becoming clear now that many of the pro-atherogenic effects demonstrated in vitro in initial studies could have been due to the presence of pro-inflammatory substances contaminating commercially sourced recombinant CRP from bacterial sources, or to the sodium azide preservative.62–70 More recent experimental studies, using pure CRP preparations, have failed to confirm the proposed pro-atherogenic actions of CRP on vascular tissues and would be concordant with our own data from genetic epidemiology,62–70 as would the recent experiments in atherosclerosis-prone mice, which showed that in contrast to earlier studies transgenic overexpression of human CRP did not enhance development of atherosclerotic lesions.71–73
In conclusion, Caucasian men with a genotype that would have exposed them to a long-term elevation in CRP concentration [0.68 mg/l (95% CI 0.31–1.10)] were not at increased risk of non-fatal MI (OR 1.01; 95% CI 0.74–1.38), though the confidence limits currently encompass substantial uncertainty. Taking this into account, it is at the very least likely that unbiased and non-confounded estimates of the effect of CRP on coronary events are smaller than earlier studies estimated, a finding that is in agreement with the latest evidence derived from prospective observational studies and also with more refined and carefully conducted mechanistic studies.74 If our null point estimate of the genetic effect is stable with the addition of further studies, it might lead to the re-evaluation of CRP both as a risk marker4 and as a potential therapeutic target.5 Additional studies with data on CRP genotype, CRP levels, and coronary events will be essential to obtain more precise genetic risk estimates and to help clarify whether or not CRP plays a causal role in atherothrombosis.
Declaration of interest
The authors have declared no conflict of interest.
Both authors contributed equally to this article.
This work was partially supported by a BHF-PhD studentship [FS/02/086/14760] awarded to T.S. J.W.S. was supported by Diabetes UK. A.D.H. holds a British Heart Foundation Senior Fellowship. S.H. holds the British Heart Foundation Chair of Cardiovascular Genetics (RG 2000/015). The HIFMECH study was supported by the Commission of the European Community: Hifmech Study contract BMH4-CT96-0272. The LEADER trial was supported by grants from the British Heart Foundation and the Medical Research Council. The NPHS-II study was supported by the British Medical Research Council, the US National Institute of Health (NHLBI 33014), and DuPont Pharma, Wilmington, USA. The WOSCOPS genotype analysis was partly funded by a research grant from Chest, Heart and Stroke Scotland. The different funding organizations named above had no role in the design and conduct, data management and analysis, manuscript preparation, or in the review or authorization for submission. The authors acknowledge the critical reading and suggestions of Prof George Davey-Smith.
References
Ridker PM, Cushman M, Stampfer MJ, Tracy RP, Hennekens CH. Inflammation, aspirin, and the risk of cardiovascular disease in apparently healthy men.
Ridker PM, Hennekens CH, Buring JE, Rifai N. C-reactive protein and other markers of inflammation in the prediction of cardiovascular disease in women.
Kuller LH, Tracy RP, Shaten J, Meilahn EN. Relation of C-reactive protein and coronary heart disease in the MRFIT nested case–control study. Multiple Risk Factor Intervention Trial.
Pearson TA, Mensah GA, Alexander RW, et al. Markers of inflammation and cardiovascular disease: application to clinical and public health practice: A statement for healthcare professionals from the Centers for Disease Control and Prevention and the American Heart Association.
Danesh J, Whincup P, Walker M, et al. Low grade inflammation and coronary heart disease: prospective study and updated meta-analyses.
Freeman DJ, Norrie J, Caslake MJ, Gaw A, Ford I, Lowe GD et al. West of Scotland Coronary Prevention Study. C-reactive protein is an independent predictor of risk for the development of diabetes mellitus in the West of Scotland Coronary Prevention Study.
Sattar N, McConnachie A, O'Reilly D, Upton MN, Greer IA, Davey Smith G et al. Inverse association between birthweight and C-reactive protein concentrations in the MIDSPAN family study.
Balk EM, Lau J, Goudas LC, Jordan HS, Kupelnick B, Kim LU et al. Effects of statins on nonlipid serum markers associated with cardiovascular disease: a systematic review.
Davey Smith G, Ebrahim S. ‘Mendelian randomization’: can genetic epidemiology contribute to understanding environmental determinants of disease?
Keavney B. Genetic epidemiological studies of coronary heart disease.
Klerk M, Verhoef P, Clarke R, Blom HJ, Kok FJ, Schouten EG et al. MTHFR 677C→T polymorphism and risk of coronary heart disease: a meta-analysis.
Wald DS, Law M, Morris JK. Homocysteine and cardiovascular disease: evidence on causality from a meta-analysis.
Casas JP, Bautista LE, Smeeth L, Sharma P, Hingorani AD. Homocysteine and stroke: evidence on a causal link from Mendelian randomisation.
Davey Smith G, Harbord R, Milton J, Ebrahim S, Sterne JA. Does elevated plasma fibrinogen increase the risk of coronary heart disease? Evidence from a meta-analysis of genetic association studies.
MacGregor AJ, Gallimore JR, Spector TD, Pepys MB. Genetic effects on baseline values of C-reactive protein and serum amyloid a protein: a comparison of monozygotic and dizygotic twins.
Pankow JS, Folsom AR, Cushman M, Borecki IB, Hopkins PN, Eckfeldt JH et al. Familial and genetic determinants of systemic markers of inflammation: the NHLBI family heart study.
D'Aiuto F, Casas JP, Shah T, Humphries SE, Hingorani AD, Tonetti M. C-reactive protein (+1444C>T) polymorphism is associated with enhanced acute release of inflammatory markers.
Brull DJ, Serrano N, Zito F, Jones L, Montgomery HE, Rumley A et al. A polymorphism in the human CRP gene influences basal and stimulated CRP levels: implications for the prediction and pathogenesis of coronary heart disease.
Miller GJ, Bauer KA, Barzegar S, Foley AJ, Mitchell JP, Cooper JA et al. The effects of quality and timing of venepuncture on markers of blood coagulation in healthy middle-aged men.
The WOSCOPS Group. Baseline risk factors and their association with outcome in the West of Scotland Coronary Prevention Study. The West of Scotland Coronary Prevention Study Group.
Meade T, Zuhrie R, Cook C, Cooper J on behalf of MRC General Practice Research Framework. Bezafibrate in men with lower extremity arterial disease: randomised controlled trial.
Juhan-Vague I, Morange PE, Aubert H, Henry M, Aillaud MF, Alessi MC et al. Plasma thrombin-activatable fibrinolysis inhibitor antigen concentration and genotype in relation to myocardial infarction in the north and south of Europe.
Colhoun HM, Rubens MB, Underwood SR, Fuller JH. The effect of type 1 diabetes mellitus on the gender difference in coronary artery calcification.
Dhamrait SS, Stephens JW, Cooper JA, Acharya J, Mani AR, Moore K et al. Cardiovascular risk in healthy men and markers of oxidative stress in Diabetics men are associated with common variation in the gene for uncoupling protein 2.
Pearson TA, Blair SN, Daniels SR, Eckel RH, Fair JM, Fortmann SP et al. AHA Guidelines for Primary Prevention of Cardiovascular Disease and Stroke: 2002 Update: Consensus Panel Guide to Comprehensive Risk Reduction for Adult Patients Without Coronary or Other Atherosclerotic Vascular Diseases. American Heart Association Science Advisory and Coordinating Committee.
Tunstall-Pedoe H, Kuulasmaa K, Amouyel P, Arveiler D, Rajakangas AM, Pajak A. Myocardial infarction and coronary deaths in the World Health Organization MONICA Project. Registration procedures, event rates, and case-fatality rates in 38 populations from 21 countries in four continents.
Mantel N, Haenszel W. Statistical aspects of the analysis of data from retrospective studies of disease.
Shadish WR, Haddock CK. Combining estimates of effect size. In: Cooper H, Hedges LV (eds). The Handbook of Research Synthesis. New York: Rusell Sage Foundation,
Deeks JJ, Altman DG, Bradburn MJ. Statistical methods for examining heterogeneity and combining results from several studies in a meta-analysis. In: Egger M, Davey Smith G, Altman DG (eds). Systematic Reviews in Health Care: Meta-Analysis in Context. London: BMJ Publications,
Danesh J, Wheeler JG, Hirschfield GM, Eda S, Eiriksdottir G, Rumley A et al. C-reactive protein and other circulating markers of inflammation in the prediction of coronary heart disease.
Altman DG, Bland JM. Interaction revisited: the difference between two estimates.
Russell AI, Cunninghame Graham DS, Shepherd C, Roberton CA, Whittaker J, Meeks J et al. Polymorphism at the C-reactive protein locus influences gene expression and predisposes to systemic lupus erythematosus.
Kovacs A, Green F, Hansson LO, Lundman P, Samnegard A, Boquist S et al. A novel common single nucleotide polymorphism in the promoter region of the C-reactive protein gene associated with the plasma concentration of C-reactive protein.
Miller DT, Zee RY, Suk Danik J, Kozlowski P, Chasman DI, Lazarus R et al. Association of common CRP gene variants with CRP levels and cardiovascular events.
Carlson CS, Aldred SF, Lee PK, Tracy RP, Schwartz SM, Rieder M et al. Polymorphisms within the C-reactive protein (CRP) promoter region are associated with plasma CRP levels.
Davey Smith G, Lawlor DA, Harbord R, et al. Association of C-reactive protein with blood pressure and hypertension. Life course confounding and Mendelian randomization tests of causality.
Woodward M, Rumley A, Lowe GD, Tunstall-Pedoe H. C-reactive protein: associations with haematological variables, cardiovascular risk factors and prevalent cardiovascular disease.
Hackam DG, Anand SS. Emerging risk factors for atherosclerotic vascular disease: a critical review of the evidence.
Primatesta P, Falaschetti E, Poulter NR. Influence of hormone replacement therapy on C-reactive protein: population-based data.
Bautista LE. Inflammation, endothelial dysfunction, and the risk of high blood pressure: epidemiologic and biological evidence.
Saito M, Ishimitsu T, Minami J, Ono H, Ohrui M, Matsuoka H. Relations of plasma high-sensitivity C-reactive protein to traditional cardiovascular risk factors.
Visser M, Bouter LM, McQuillan GM, Wener MH, Harris TB. Elevated C-reactive protein levels in overweight and obese adults.
Stewart SH, Mainous AG III, Gilbert G. Relation between alcohol consumption and C-reactive protein levels in the adult US population.
Bautista LE, Smeeth L, Hingorani AD, Casas JP. Estimation of bias in non-genetic observational studies using ‘Mendelian triangulation’.
Davey Smith G, Harbord R, Ebrahim S. Fibrinogen, C-reactive protein and coronary heart disease: does Mendelian randomization suggest the associations are non-causal?
Zee RY, Ridker PM. Polymorphism in the human C-reactive protein (CRP) gene, plasma concentrations of CRP, and the risk of future arterial thrombosis.
KWJ Lee, J Hill, J Frohlich. C-reactive protein as a predictor of clinical outcomes in patients with coronary atherosclerosis. (Oral session young investigator award). Canadian Cardiovascular Congress,
Goldstein DB, Ahmadi KR, Weale ME, Wood NW. Genome scans and candidate gene approaches in the study of common diseases and variable drug responses.
SeattleSNPs. NHLBI Program for Genomic Applications, SeattleSNPs, Seattle, WA. (http://pga.gs.washington.edu) (Accessed May 1,
Szalai AJ, Wu J, Lange EM, McCrory MA, Langefeld CD, Williams A et al. Single-nucleotide polymorphisms in the C-reactive protein (CRP) gene promoter that affect transcription factor binding, alter transcriptional activity, and associate with differences in baseline serum CRP level.
Timpson NJ, Lawlor DA, Harbord RM, Gaunt TR, Day IN, Palmer LJ et al. C-reactive protein and its role in metabolic syndrome: mendelian randomisation study.
Verma S, Kuliszewski MA, Li SH, Szmitko PE, Zucco L, Wang CH et al. C-reactive protein attenuates endothelial progenitor cell survival, differentiation, and function: further evidence of a mechanistic link between C-reactive protein and cardiovascular disease.
Li SH, Szmitko PE, Weisel RD, Wang CH, Fedak PW, Li RK et al. C-reactive protein upregulates complement-inhibitory factors in endothelial cells.
Wang CH, Li SH, Weisel RD, Fedak PW, Dumont AS, Szmitko P et al. C-reactive protein upregulates angiotensin type 1 receptors in vascular smooth muscle.
Verma S, Wang CH, Li SH, Dumont AS, Fedak PW, Badiwala MV et al. A self-fulfilling prophecy: C-reactive protein attenuates nitric oxide production and inhibits angiogenesis.
Pasceri V, Willerson JT, Yeh ET. Direct proinflammatory effect of C-reactive protein on human endothelial cells.
Pasceri V, Cheng JS, Willerson JT, Yeh ET. Modulation of C-reactive protein-mediated monocyte chemoattractant protein-1 induction in human endothelial cells by anti-atherosclerosis drugs.
Taylor KE, Giddings JC, van den Berg CW. C-Reactive protein-induced in vitro endothelial cell activation is an artefact caused by azide and lipopolysaccharide.
Lafuente N, Azcutia V, Matesanz N, Cercas E, Rodriguez-Manas L, Sanchez-Ferrer CF et al. Evidence for sodium azide as an artifact mediating the modulation of inducible nitric oxide synthase by C-reactive protein.
van den Berg CW, Taylor KE, Lang D. C-reactive protein-induced in vitro vasorelaxation is an artefact caused by the presence of sodium azide in commercial preparations.
Swafford AN Jr, Bratz IN, Knudson JD, Rogers PA, Timmerman JM, Tune JD et al. C-reactive protein does not relax vascular smooth muscle: effects mediated by sodium azide in commercially available preparations.
Clapp BR, Hirschfield GM, Storry C, Gallimore JR, Stidwill RP, Singer M et al. Inflammation and endothelial function: direct vascular effects of human C-reactive protein on nitric oxide bioavailability.
van den Berg CW, Taylor KE. Letter Regarding Article by Li et al, “C-Reactive Protein Upregulates Complement-Inhibitory Factors in Endothelial Cells”.
Liu C, Wang S, Deb A, Nath KA, Katusic ZS, McConnell JP et al. Proapoptotic, antimigratory, antiproliferative, and antiangiogenic effects of commercial C-reactive protein on various human endothelial cell types in vitro: implications of contaminating presence of sodium azide in commercial preparation.
Nerurkar SS, McDevitt PJ, Scott GF, Johanson KO, Willette RN, Yue TL. Lipopolysaccharide (LPS) contamination plays the real role in C-reactive protein-induced IL-6 secretion from human endothelial cells in vitro.
Pepys MB, Hawkins PN, Kahan MC, Tennent GA, Gallimore JR, Graham D et al. Proinflammatory effects of bacterial recombinant human C-reactive protein are caused by contamination with bacterial products, not by C-reactive protein itself.
Hirschfield GM, Gallimore JR, Kahan MC, Hutchinson WL, Sabin CA, Benson GM et al. Transgenic human C-reactive protein is not proatherogenic in apolipoprotein E-deficient mice.
Reifenberg K, Lehr H-A, Baskal D, Wiese E, Schaefer SC, Black S et al. Role of C-reactive protein in atherogenesis. Can the apolipoprotein E knockout mouse provide the answer?
Trion A, de Maat MP, Jukema JW, van der Laarse A, Maas MC, Offerman EH et al. No effect of C-reactive protein on early atherosclerosis development in apolipoprotein E*3-Leiden/human C-reactive protein transgenic mice.


{kind=link}
{kind=link}
{kind=link}