





Abstract
Aims Non-compaction of the left ventricle (LVNC) is a disorder of endomyocardial morphogenesis that results in multiple trabeculations in the left ventricular myocardium. The current literature suggests that LVNC in adults is rare and associated with a poor prognosis. Given that the disorder is present at birth and that several studies have reported asymptomatic familial disease in some patients, we hypothesized that there is a long pre-clinical phase of the disease. The aim of this study was to define the prognosis and familial incidence of LVNC.
Methods and results This study cohort comprised 45 patients (mean age at diagnosis 37 years) consecutively identified at a referral centre for cardiomyopathy over a 10-year period. Twenty-eight patients (62%) had dyspnoea at presentation; 41 (91%) an abnormal ECG; and 30 (66%) left ventricular dilatation and impaired systolic function. Nine patients (20%) had non-sustained ventricular tachycardia on 24 h Holter monitoring. Mean survival from death or transplantation was 97% at 46 months. There were three thromboembolic events in two patients (4%). On systematic family screening, 8 of 32 (25%) asymptomatic relatives had a range of echocardiographic abnormalities, including LVNC, LVNC with impaired systolic function, and left ventricular enlargement without LVNC.
Conclusion This study demonstrates that LVNC is associated with a better prognosis than previously reported. In patients with familial disease, relatives may have features consistent with dilated cardiomyopathy rather than LVNC.
Introduction
Non-compaction of the left ventricle (LVNC) is a disorder of endomyocardial morphogenesis that results in multiple trabeculations in the left ventricular myocardium.1 The current literature suggests that LVNC in adults is rare, and associated with a poor prognosis.2–5 However, many studies have reported on highly symptomatic cases with a high incidence of ventricular arrhythmia and progressive heart failure. Given that the disorder is present at birth and that several studies have reported asymptomatic familial disease in some patients, we hypothesized that there is a long pre-clinical phase of disease and that the published prognosis figures are unrepresentative of the true natural history of the disease.
Methods
Patients
The study cohort comprised 45 unrelated patients with LVNC consecutively referred to the cardiomyopathy clinic at St George's Hospital, London, UK, between January 1992 and June 2002. The reason for referral was for diagnosis clarification (n=16), following a thromboembolic event (n=2), family screening for dilated cardiomyopathy (n=2), management of heart failure (n=25). In the same period, 518 patients with idiopathic dilated cardiomyopathy (DCM) (impaired systolic function and left ventricular dilatation in the absence of a definable cause) were reviewed in the same clinic. All patients underwent systematic clinical evaluation, including history and examination, electrocardiogram (ECG), two-dimensional and Doppler echocardiography, metabolic exercise testing, and 24 h Holter monitoring. Clinical and echocardiographic follow-up in the cardiomyopathy clinic was performed in all patients at intervals of at least 6 months. Detailed pedigree analysis was undertaken prior to outpatient physician assessment in which a trained research nurse specializing in pedigree analysis interviewed the patient and his/her family in depth. First-degree relatives of LVNC probands were offered clinical screening using the same protocol.
Echocardiography
Transthoracic two-dimensional and Doppler echocardiography were performed using a System Five or Vivid 7 echocardiograph (GE Medical Systems). Measurements of left ventricular end-diastolic dimension (LVED) and left ventricular end-systolic dimension (LVES) were obtained in accordance with the recommendations of the American Society of Echocardiography.6 Fractional shortening (FS) was calculated as (LVED–LVES/LVED)×100. Dimensions were corrected for age and body surface area according to the formula of Henry: {LVED (per cent predicted)=measured LVED/predicted LVED×100; predicted LVED=[45.3×body surface area (BSA)0.3]−(0.03×Age)–7.2}.7 Left ventricular abnormalities were classified as follows: DCM, LVED ≥ 117% predicted and fractional shortening (FS) < 25% in the absence of known causes of ventricular dilatation;7,8 left ventricular enlargement (LVE), LVED ≥ 112% predicted, with normal systolic function.9
Diagnostic criteria for LVNC
Echocardiographic criteria for LVNC have been described by Chin et al.1 These focus on trabeculae at the LV apex on the parasternal short axis and apical views, and on left ventricular free-wall thickness at end-diastole. LVNC is defined by a ratio of X/Y≤0.5 where X is the distance from the epicardial surface to the trough of the trabecular recess, and Y is the distance from the epicardial surface to peak of trabeculation.
Another proposed standardized method of identifying LVNC10 is based on:
absence of co-existing cardiac structural abnormalities;
numerous, excessively prominent trabeculations and deep intratrabecular recesses;
recesses supplied by intraventricular blood on colour Doppler; and
a two-layer structure, with thin compacted layer and a thick non-compacted layer.
End-systolic thickness of the compacted layer and the non-compacted layer of endomyocardium is taken at maximal thickness to calculate the ratio of non-compacted/compacted (N/C) at parasternal short axis views; LVNC is defined where N/C > 2.
Cardiopulmonary exercise testing
Patients underwent metabolic cardiopulmonary exercise testing on a bicycle ergometer (Sensormedics Ergometrics 800S) using a ramp protocol of 10–15 W with respiratory gas sampling (V Max 29 Console, Sensormedics) and serial measurements of blood pressure during exercise. Peak oxygen consumption (peak VO2) was defined as the highest VO2 achieved during exercise. Results were expressed as a percentage of the predicted maximal VO2 to allow for age, gender, and body size, and a value of <80% of predicted was considered a marker of significant impairment.
Statistical analysis
Descriptive data for continuous variables are presented as mean ± standard deviation. Continuous data were compared using paired and unpaired Student's t-test and χ2 analysis where appropriate. A SPSS v10.0 (Chicago, IL, USA) statistical package was used. A P-value<0.05 was considered significant.
Results
Baseline characteristics
Clinical, ECG, and echocardiographic characteristics of the 45 patients are presented in Table 1. LVNC was the primary diagnosis in 25 patients. The initial diagnosis in the remaining 20 patients had been DCM (n=18) and hypertrophic cardiomyopathy (n=2). The reasons for reclassification were missed diagnosis at referral (n=14) or revision of diagnosis at our own centre (n=6), due to improved imaging techniques and increased awareness by our echocardiographers. LVNC was diagnosed on the basis of one or both published standard criteria: 32 patients fulfilled Chin criteria1 for LVNC, 41 fulfilled Jenni criteria10 and 22 fulfilled both. Twenty patients (44%) had predominantly apical LVNC. No patient had concomitant congenital heart disease, clinical evidence of skeletal myopathy, or facial dysmorphism.
Thirty (67%) patients had left ventricular dilatation and impaired systolic function; in these patients, the NYHA class was 2.3±0.76 (mean±SD) at presentation, and the FS was 15±6%. Fifteen (33%) patients had LVNC with normal systolic function (FS 31±4%); all of these patients were in NYHA functional class 1. Two patients with LVNC and normal systolic function had left ventricular enlargement. Twenty-three (51%) patients had a family history of DCM and/or LVNC.
Forty-one (91%) patients had an abnormal ECG. Abnormalities included left bundle branch block (LBBB) (n=13, 29%), poor R wave progression (n=3, 7%), ST-segment change (n=4, 9%), pathological Q waves (n=4, 9%), and T wave inversion (n=7, 16%). Three patients were in atrial fibrillation at the time of diagnosis.
Peak predicted VO2 achieved was 67±18% (mean±SD) in the LVNC and impaired systolic function group vs. 86±21% in the LVNC and normal systolic function group (P=0.03). One patient, with normal systolic function, had five beats of non-sustained ventricular tachycardia at peak exercise.
Follow-up
Of those patients with LVNC and impaired systolic function (n=30), 18 (60%) received long-term anticoagulation; 27 patients (90%) were on angiotensin converting enzyme (ACE-) inhibitors or angiotensin receptor blockers; 14 (47%) received beta-blockers, and two (7%) patients received amiodarone. Three patients received biventricular pacemakers (InSynch III, Medtronic, Minneapolis, MN, USA) for symptomatic heart failure. Of the 15 patients with LVNC and normal systolic function, two (13%) were receiving ACE-inhibitors and anticoagulation; one with left ventricular enlargement; the other had presented with a transient ischaemic attack at the age of 19.
Nine patients (20%) had non-sustained ventricular arrhythmias on 24 h Holter monitoring, eight of whom had impaired systolic function at baseline. Two patients received implantable cardioverter defibrillators (ICDs) for primary prevention; neither patient had an ICD discharge.
During a mean follow-up of 46 months (median 32, range 6–179), mean NYHA class in the LVNC and impaired systolic function group (n=30) improved from 2.3±0.6 to 1.4±0.4 (P=0.002). FS did not change in this group (15±6 to 17±5%, P=0.8). In patients with LVNC and normal systolic function at baseline, there was no death or progression to heart failure.
During a mean follow-up of 46 months (median 32, range 6–179), there was one sudden cardiac death in a 28-year-old proband with NYHA class III heart failure awaiting cardiac transplant. During 10 years of follow-up there were three thromboembolic episodes in two patients (4%), both of whom were in sinus rhythm. One patient, with impaired LV function, who had fully recovered from a thromboembolic stroke, had another episode 10 years later, despite anticoagulation.
Family screening
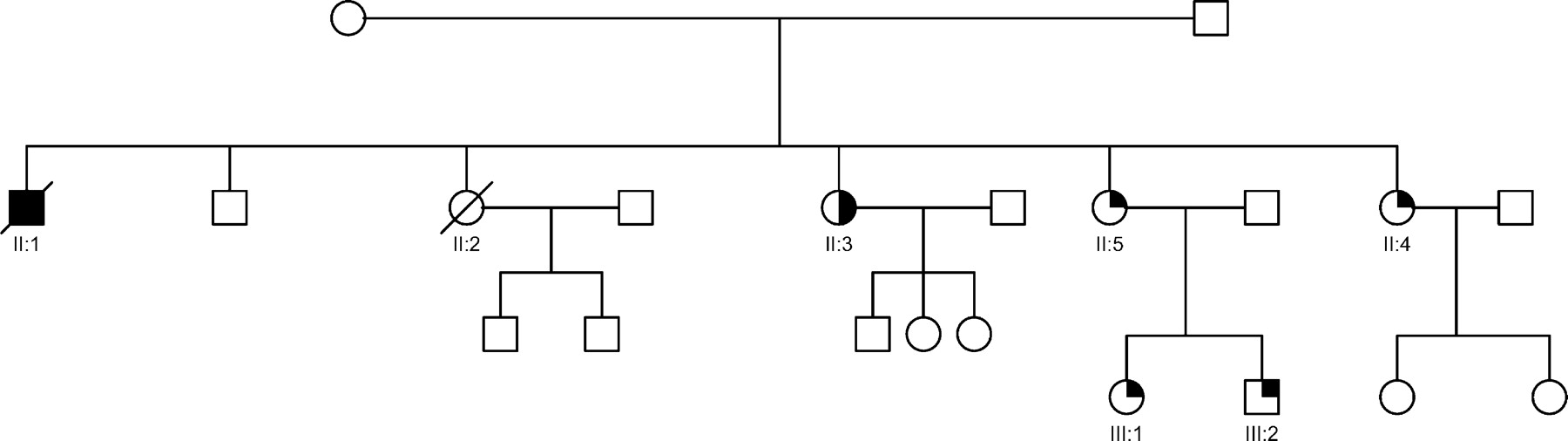
Twenty-two (49%) of the 45 families consented to systematic screening. Thirty-two asymptomatic first-degree relatives, from a total of 57 (56%), in these 22 families were available for assessment. A sample family pedigree is shown in Figure 1. Of these 22 families, 16 had a family history of cardiomyopathy or sudden death. Of the 32 relatives screened, eight asymptomatic individuals (25%) were found to have echocardiographic abnormalities (see Figures 2 and 3). Two of the six families with no history of cardiomyopathy or sudden death had relatives with LVNC on echocardiographic screening. There was a range of abnormalities found: LVNC and normal systolic function in four, LVNC with impaired systolic function in two, left ventricular enlargement (with LVED of 118% predicted) in one, and a thickened slightly trabeculated posterior wall with LVE in another subject, which did not fulfil criteria for LVNC. This individual developed syncope, had frequent ventricular ectopics and runs of non-sustained VT on Holter monitoring, and received an ICD. At follow-up, all eight patients remained in NYHA class I, with no deterioration in systolic function. The uncle of one LVNC proband (Figure 1, II : 1) had received a cardiac transplant for refractory heart failure secondary to DCM. A review of the histology on the explanted heart was available and showed dilated ventricles, and extensive diffuse interstitial fibrosis without evidence of LVNC or excessive trabeculations.
Discussion
This study shows that patients with LVNC have a more favourable prognosis then previously described.2,3 It also shows that familial LVNC encompasses a spectrum of abnormalities that overlaps with that seen in families with DCM, suggesting that in some cases the diseases may share a common aetiology.
Pathogenesis of non-compaction
During normal embryonic development, endomyocardial trabeculations emerge from the apical region of the primitive ventricles at day 32 of foetal life,11 and involute by day 70 through a process of resorption and remodelling. LVNC is thought to represent a failure of this ‘compaction’ process. In children, LVNC can occur in Barth syndrome, a rare X-linked multi-system disorder caused by a mutation in the G4.5 gene that encodes the tafazzin family of proteins.12,13 However, mutations in this gene in adult LVNC are rare and no mutations in this gene were identified in the first 15 of the 45 patients included in this study.14,15 Mutations have been described in adult LVNC in the genes encoding α-dystrobrevin and Cypher/ZASP,12,16 integral parts of the complex which links the extracellular matrix of the myocardial cell to the cytoskeleton and two patients in the current study carried the D117N mutation in Cypher/ZASP.16 The fact that Cypher/ZASP and α-dystrobrevin are important structural proteins is intriguing as most published mutations in families with DCM also affect components of the cellular cytoskeleton.17,18 This common link may explain the frequent development of ventricular dilatation and systolic impairment in patients with LVNC, and may also explain the occurrence of DCM and LVE without LVNC in relatives of LVNC patients.
Natural history
Most detailed clinical descriptions of LVNC in the literature have suggested that patients with LVNC have poorly functioning dilated ventricles, a high incidence of ventricular arrhythmias, and systemic emboli. In the largest series to date, 48% of patients died or underwent cardiac transplantation over a period of 44 months.2 In contrast, many of the patients identified in this study had a milder phenotype with a much lower incidence of death, stroke, or documented sustained ventricular arrhythmia. This may be explained partly by patient selection and screening. Improvements in echocardiographic imaging may also have facilitated detection of previously unrecognized asymptomatic cases. The effect of therapeutic intervention in asymptomatic cases is difficult to determine from this small study. Similarly, there were insufficient clinical events to show a significant difference in outcome in those patients treated with anticoagulants, ACE-inhibitors, beta-blockers, or amiodarone. However, the study does support the use of anticoagulation in LVNC, particularly where there is impaired systolic function (FS < 25%), or a history of thromboembolism. The occurrence of a transient ischaemic attack in one patient with normal systolic function and dense apical LVNC suggests that dense and extensive non-compaction alone may be an indication for anticoagulation.
Clinical implications
This study has several important implications for clinical practice. First, the fact that LVNC was often detected retrospectively in patients who had already been diagnosed with DCM suggests that its frequency in the heart failure population may have been underestimated, probably as the result of inadequate imaging of the apical segments of the left ventricle. Lack of physician awareness may also have led to an underestimation of the incidence of LVNC. This is in keeping with recent reviews which noted an increased incidence of LVNC with improving cardiac imaging.19 Of the two published criteria for LVNC available, Jenni criteria10 stress the presence of a two-layered structure, whereas Chin criteria1 focus on the depth of recess compared with the height of trabeculae. These differences may explain the fact that some patients fulfilled only one of the two diagnostic criteria. For example, it may not always be possible to resolve the two-layered appearance on two-dimensional echo. Another important difference is the use of end-diastolic measurements in the Chin method compared with end-systolic measurements in Jenni. The fact that only 22 patients fulfilled both Jenni and Chin definitions of LVNC, suggests that they are complementary in diagnosing LVNC.
Second, we have shown previously that relatives of DCM patients have subtle abnormalities including LVE,9 diminished metabolic exercise capacity,20 and abnormal histology;21 25% of such cases progress to symptomatic DCM. Systematic family screening in this study revealed LVE and asymptomatic LVNC in relatives of LVNC patients, which implies a similar long period of silent gestation before the onset of clinical disease. Moreover, the presence of DCM and LVE without LVNC in relatives of LVNC patients shows that the phenotype of familial LVNC can overlap with that seen in familial DCM. These data suggest that isolated LVNC could be classified as a sub-type or variant of idiopathic DCM rather than a distinct cardiomyopathy in itself.
Acknowledgements
R.T.M. was supported by the Irish Heart Foundation/Novartis, R.T., and W.J.McK. by the British Heart Foundation.
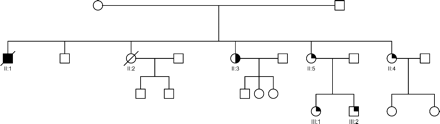
Figure 1 Phenotypic heterogeneity in LVNC. Family pedigree NC3 showing phenotypic heterogeneity, with isolated DCM, isolated LVNC and LVE co-existing within the same family. The family was screened after the death of a proband (II : 1) with DCM, who died 3 years after a cardiac transplant for end-stage heart failure. His explanted heart showed DCM and on review had no evidence of LVNC or excessive trabeculations of the left ventricle. A sister (II : 2) died of cardiac failure immediately post-partum. Another sister (II : 3) had a mildly dilated left ventricle, and systolic function at the lower limit of normal (FS=26%). A further sister (II : 4) had a history of syncope, isolated left ventricular enlargement, a thickened posterior wall, preserved systolic function, frequent runs of non-sustained ventricular tachycardia on cardiac monitoring, and received a prophylactic ICD. Another sister (II : 5), her daughter (III : 1), and son (III : 2), have extensive LVNC and normal systolic function. Solid square and circle symbols indicate affected males and females with DCM, respectively; open symbols, unaffected; half-symbols, left ventricular enlargement; quarter symbols, LVNC; and slashes, death.
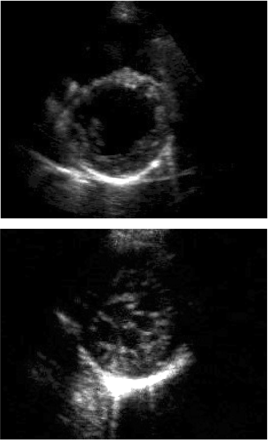
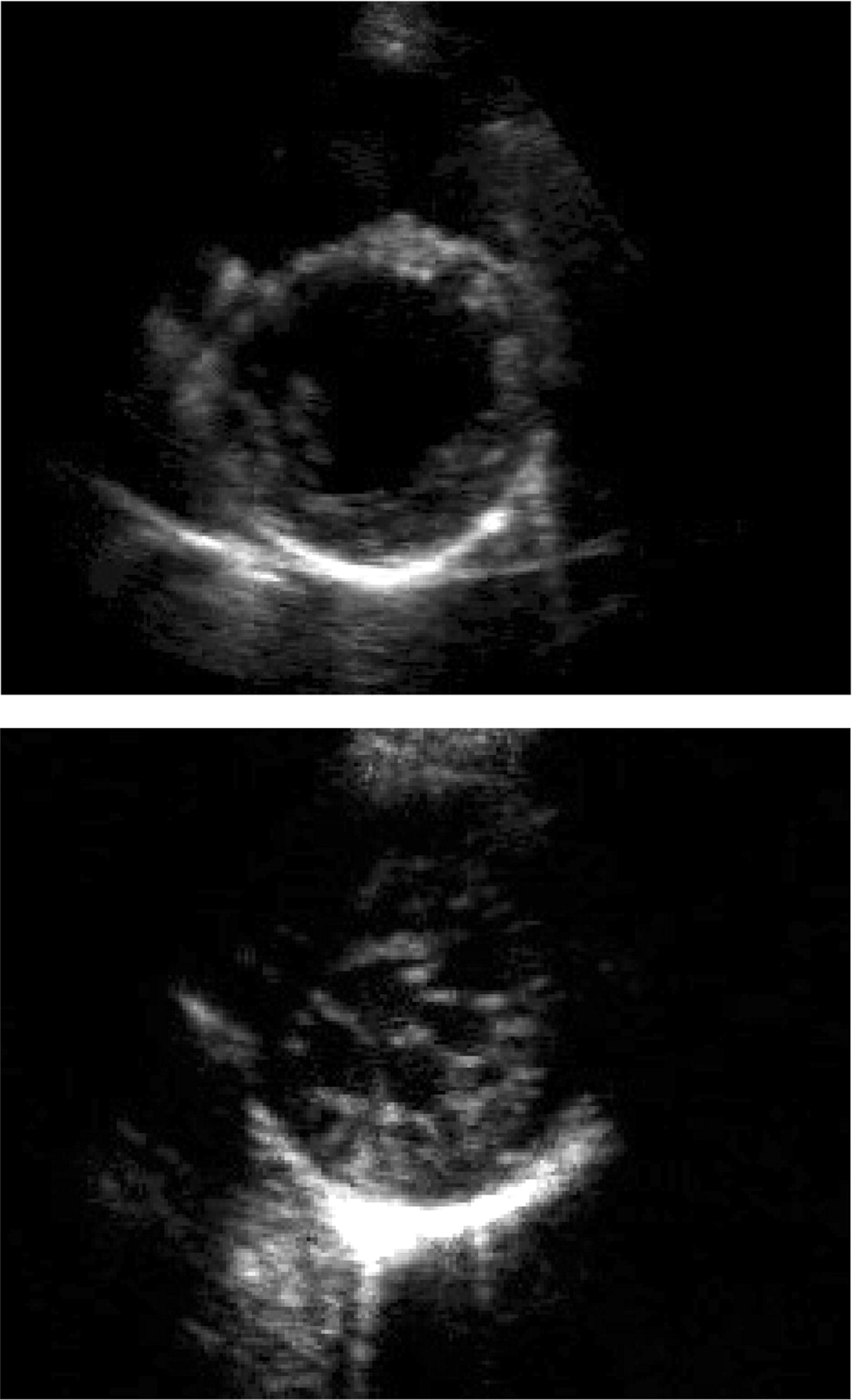
Figure 2 Parasternal short axis views (end-diastole) of a 34-year-old patient (II : 4 in Figure 1) with left ventricular enlargement (LVED 118% predicted) and minimal posterolateral trabeculations (upper), and her 12-year-old niece (III:1) with an extensive meshwork of trabeculations and recesses communicating with the LV cavity (lower).
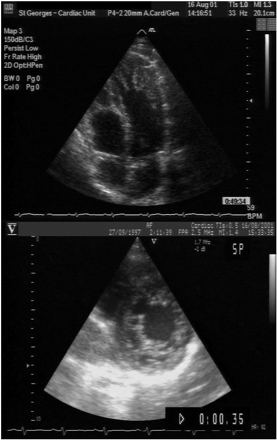
Figure 3 Apical four-chamber view (end-diastole) of a 36-year-old patient (upper) referred with an initial diagnosis of apical hypertrophic cardiomyopathy, with extensive apical trabeculations and recesses consistent with LVNC, and (lower) a parasternal short-axis view (end-diastole) of his asymptomatic 4-year-old son, showing a two-layered structure of compacted and non-compacted endomyocardium. Colour Doppler showed flow between the LV cavity and the recesses.
Clinical and echocardiographic characteristics of LVNC probands (n=45)
| Male/female | 28/17 |
| Age at diagnosis, yearsa | 37±17 (13–83) |
| NYHA classb | |
| I | 16 (36) |
| II | 13 (28) |
| III/IV | 16 (36) |
| NSVT on Holter monitoringb | 10 (22) |
| Chest painb | 6 (13) |
| Pre-syncope/syncopeb | 4 (9) |
| Family history DCM/LVNCb | 23 (51) |
| Abnormal ECGb | 41 (91) |
| Fractional shortening,%a | 21±9 (5–39) |
| LVED, mma | 58±11 (37–94) |
| LVES, mma | 46±14 (22–88) |
| LAD, mma | 40±8 (22–58) |
| Median peak VO2 mL/kg/min | 19.5 (range 12.2–52) |
| Mean peak predicted VO2, % | 74 (range 45–127) |
| Male/female | 28/17 |
| Age at diagnosis, yearsa | 37±17 (13–83) |
| NYHA classb | |
| I | 16 (36) |
| II | 13 (28) |
| III/IV | 16 (36) |
| NSVT on Holter monitoringb | 10 (22) |
| Chest painb | 6 (13) |
| Pre-syncope/syncopeb | 4 (9) |
| Family history DCM/LVNCb | 23 (51) |
| Abnormal ECGb | 41 (91) |
| Fractional shortening,%a | 21±9 (5–39) |
| LVED, mma | 58±11 (37–94) |
| LVES, mma | 46±14 (22–88) |
| LAD, mma | 40±8 (22–58) |
| Median peak VO2 mL/kg/min | 19.5 (range 12.2–52) |
| Mean peak predicted VO2, % | 74 (range 45–127) |
NSVT, non-sustained ventricular tachycardia > 3 beats on 48 h Holter monitoring; LAD, left atrial diameter; NYHA class, New York Heart Association class (at presentation).
a Mean±SD (range).
bn (%).
Clinical and echocardiographic characteristics of LVNC probands (n=45)
| Male/female | 28/17 |
| Age at diagnosis, yearsa | 37±17 (13–83) |
| NYHA classb | |
| I | 16 (36) |
| II | 13 (28) |
| III/IV | 16 (36) |
| NSVT on Holter monitoringb | 10 (22) |
| Chest painb | 6 (13) |
| Pre-syncope/syncopeb | 4 (9) |
| Family history DCM/LVNCb | 23 (51) |
| Abnormal ECGb | 41 (91) |
| Fractional shortening,%a | 21±9 (5–39) |
| LVED, mma | 58±11 (37–94) |
| LVES, mma | 46±14 (22–88) |
| LAD, mma | 40±8 (22–58) |
| Median peak VO2 mL/kg/min | 19.5 (range 12.2–52) |
| Mean peak predicted VO2, % | 74 (range 45–127) |
| Male/female | 28/17 |
| Age at diagnosis, yearsa | 37±17 (13–83) |
| NYHA classb | |
| I | 16 (36) |
| II | 13 (28) |
| III/IV | 16 (36) |
| NSVT on Holter monitoringb | 10 (22) |
| Chest painb | 6 (13) |
| Pre-syncope/syncopeb | 4 (9) |
| Family history DCM/LVNCb | 23 (51) |
| Abnormal ECGb | 41 (91) |
| Fractional shortening,%a | 21±9 (5–39) |
| LVED, mma | 58±11 (37–94) |
| LVES, mma | 46±14 (22–88) |
| LAD, mma | 40±8 (22–58) |
| Median peak VO2 mL/kg/min | 19.5 (range 12.2–52) |
| Mean peak predicted VO2, % | 74 (range 45–127) |
NSVT, non-sustained ventricular tachycardia > 3 beats on 48 h Holter monitoring; LAD, left atrial diameter; NYHA class, New York Heart Association class (at presentation).
a Mean±SD (range).
bn (%).
References
Chin TK, Perloff JK, Williams RG et al. Isolated noncompaction of left ventricular myocardium: a study of eight cases.
Oechslin EN, Attenhofer Jost CH, Rojas JR et al. Long-term follow-up of 34 adults with isolated left ventricular noncompaction: a distinct cardiomyopathy with poor prognosis.
Ichida F, Hamamichi Y, Miyawaki T et al. Clinical features of isolated noncompaction of the ventricular myocardium: long-term clinical course, hemodynamic properties, and genetic background.
Ritter M, Oechslin E, Sutsch G et al. Isolated noncompaction of the myocardium in adults.
Shah CP, Nagi KS, Thakur RK et al. Spongy left ventricular myocardium in an adult.
Schiller NB, Shah PM, Crawford M et al. Recommendations for quantitation of the left ventricle by two-dimensional echocardiography. American Society of Echocardiography committee on standards, subcommittee on quantitation of two-dimensional echocardiography.
Henry WL, Gardin JM, Ware JH. Echocardiographic measurements in normal subjects from infancy to old age.
Richardson P, McKenna W, Bristow M et al. Report of the 1995 World Health Organization/International Society and Federation of Cardiology Task Force on the definition and classification of cardiomyo-pathies.
Baig MK, Goldman JH, Caforio AL et al. Familial dilated cardiomyopathy: cardiac abnormalities are common in asymptomatic relatives and may represent early disease.
Jenni R, Oechslin E, Schneider J et al. Echocardiographic and patho-anatomical characteristics of isolated left ventricular non-compaction: a step towards a classification as a distinct cardiomyopathy.
Collins P. Embryology: development of the heart. In: Williams PL, ed. Gray's Anatomy, 38th ed. London: Churchill Livingstone;
Ichida F, Tsubata S, Bowles KR et al. Novel gene mutations in patients with left ventricular noncompaction or Barth syndrome.
Bleyl SB, Mumford BR, Thompson V et al. Neonatal, lethal noncompaction of the left ventricular myocardium is allelic with Barth syndrome.
Sasse-Klaassen S, Gerull B, Oechslin E et al. Isolated noncompaction of the left ventricular myocardium in the adult is an autosomal dominant disorder in the majority of patients.
Kenton AB, Sanchez X, Coveler KJ et al. Isolated left ventricular noncompaction is rarely caused by mutations in G4.5, alpha-dystrobrevin and FK Binding Protein-12.
Vatta M, Mohapatra B, Jimenez S et al. Mutations in Cypher/ZASP in patients with dilated cardiomyopathy and left ventricular non-compaction.
Shaw T, Elliott P, McKenna WJ. Dilated cardiomyopathy: a genetically heterogeneous disease.
Franz WM, Muller OJ, Katus HA. Cardiomyopathies: from genetics to the prospect of treatment.
Stollberger C, Finsterer J. Left ventricular hypertrabeculation/noncompaction.
Mahon NG, Sharma S, Elliott PM et al. Abnormal cardiopulmonary exercise variables in asymptomatic relatives of patients with dilated cardiomyopathy who have left ventricular enlargement.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}