
Abstract
Relaxin is a naturally occurring human peptide initially identified as a reproductive hormone. More recently, relaxin has been shown to play a key role in the maternal hemodynamic and renal adjustments that accommodate pregnancy. An understanding of these physiologic effects has led to the evaluation of relaxin as a pharmacologic agent for the treatment of patients with acute heart failure. Preliminary results have been encouraging. In addition, the other known biologic properties of relaxin, including anti-inflammatory effects, extracellular matrix remodeling effects, and angiogenic and anti-ischemic effects, all may play a role in potential benefits of relaxin therapy. Ongoing, large-scale clinical testing will provide additional insights into the potential role of relaxin in the treatment of heart failure.
Similar content being viewed by others

Introduction
Hospitalizations for acute heart failure (AHF) have traditionally been viewed as resulting from salt and fluid accumulation in patients with chronic heart failure. Diuretic therapy has evolved to be, and is still considered, the best intervention for acute symptom relief, despite the growing recognition of its limitations and the absence of compelling clinical data to support its use [1]. The significant contribution of vascular dysfunction to the pathophysiology of AHF has more recently been recognized [2•]. These patients are characterized by preserved or elevated systolic blood pressure and increased vascular stiffness with less fluid overload. They are more likely to be elderly and female. Large-scale registry studies suggest that patients with vascular dysfunction causing AHF represent the majority of patients, and that this may have been underappreciated during previous development of new therapies.
An even more comprehensive and integrated disease model would now include consideration of cardiac, renal, vascular, neurohormonal, inflammatory, ischemic, and fibrotic mechanisms [2•, 3]. Perhaps this growing understanding of disease complexity in AHF may lead to the development of new interventions that benefit these patients. In particular, the ability to identify different subgroups of patients with AHF may enable therapies that target the underlying pathophysiology, with the potential for greater therapeutic success.
The peptide hormone relaxin has a long history as a reproductive hormone since its discovery in 1926 [4, 5]. In women, circulatory concentrations of relaxin rise in the first trimester, coincident with notable cardiovascular and renal adjustments of the maternal circulation during pregnancy [6••]. These adjustments include systemic vasodilation to meet the increased nutritional demands of the growing fetus and increased renal function to accommodate the heightened requirements for renal clearance of metabolic wastes. These adaptations include a 20% increase in cardiac output, 30% decrease in systemic vascular resistance, 30% increase in global arterial compliance, and 45% increase in renal blood flow [6••], [7, 8]. Clinical and nonclinical studies using relaxin have recapitulated these effects in males, as well as females, leading to a growing recognition that the physiologic effects of relaxin may be pharmacologically useful in modulating cardiovascular and renal function.
This review focuses on the major biological properties of relaxin, that is, hemodynamic and renal, and their potential role in heart failure therapy. Clinical trials of relaxin in patients with heart failure have begun to generate encouraging results due to these biological properties [9••, 10••]. Evidence of relaxin pharmacology was also apparent in early clinical studies in non-heart failure indications. Other biologic properties of relaxin, including its antifibrotic, anti-inflammatory, and anti-ischemic effects, are also described, as they could potentially address the multiple facets of the pathophysiology of heart failure.
Mechanisms of Action
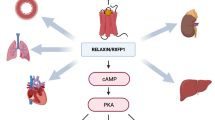
Decades of cumulative biological, pharmacologic, and clinical experimentation have led to our current understanding of the biology of relaxin, depicted in Fig. 1 [6••, 11–13••, 14]. The major effects of relaxin, consisting of the modulation of systemic and renal vascular tone, are mediated by multiple mechanisms (Fig. 1). Amongst them, its interaction with the endothelin system appears to have a pivotal role. This system is responsible, at least in part, for physiological regulation of basal vascular tone [15, 16]. Endothelin-1 (ET-1) is synthesized by vascular endothelial cells and acts through binding to specific endothelin-A (ET-A) and Endothelin-B (ET-B) receptors. ET-A receptors are located on vascular smooth muscle cells and mediate vasoconstriction. ET-B receptors on endothelial cells are vasodilatory, whereas those on smooth muscle cells are vasoconstrictive. Normal tone results from the balance between ET-1 stimulation of ET-A and ET-B receptors [16]. In heart failure, pathologically elevated levels of endothelin cause arterial and venous vasoconstriction, contribute to symptoms, and are associated with adverse outcomes [17–19]. Furthermore, in human heart failure, there is altered expression of endothelin receptors with increased ET-A receptor density and decreased ET-B receptor density [20].

Biology of relaxin and potential beneficial effects in heart failure. Heart failure is a cardiac-renal-vascular-neurohormonal-inflammatory-ischemic-fibrotic disorder. Human data indicate that relaxin relieves systemic and renal vasoconstriction and increases vascular compliance, including normalization of high BP, ↓ PCWP, ↑ CO, ↑ renal blood flow, natriuresis, and decongestion. Animal pharmacology data indicate that relaxin has anti-inflammatory and cardiac protection effects, including ↓ myocardial ischemia, ↓ reperfusion injury, ↑ wound healing, ↓ ventricular fibrosis. BP—blood pressure; CO—cardiac output; ET—endothelin; MMP—matrix metalloproteinase; NO—nitric oxide; NOS—nitric oxide synthase; PCWP—pulmonary capillary wedge pressure; TGF—transforming growth factor; TNF—tumor necrosis factor; VEGF—vascular endothelial growth factor
Relaxin activity is initiated by binding to its cognate receptor, which has been localized in the systemic and renal vasculature, as well as in the tubules of the kidney [21]. The close proximity of the relaxin receptor to components of the endothelin pathway in these anatomical locations is consistent with the perceived interaction of the two endogenously occurring systems. Furthermore, isolated vessels ex vivo respond to relaxin exposure by vasodilation, indicating that the relaxin receptor and the downstream mechanisms mediating dilation are located within the vessels themselves. Relaxin binding to its receptor results in activation of the ET-B receptor on endothelial cells, shifting the balance of vascular tone toward vasodilation [22–24]. Systemically, relaxin appears to directly stimulate expression of the ET-B receptor on endothelial cells, whereas stimulation of ET synthesis, resulting in increased engagement and activation of the endothelial ET-B receptor, appears to be the pathway in the kidney. The two pathways converge in increased local nitric oxide production. Although not as well understood, relaxin may also activate the ET-B receptor in the tubules of the kidney, thereby inhibiting the Na/K ATPase and facilitating sodium and water excretion.
Clinical Data on Vasodilation
Extensive clinical data document the vasodilatory effects of relaxin. In a dose-ranging study of relaxin given to 16 patients with stable heart failure, systemic hemodynamics were directly measured by Swan-Ganz catheterization [9••]. Vasodilation was documented as a decrease in pulmonary capillary wedge pressure (PCWP) and systemic vascular resistance (SVR), and an increase in cardiac output (CO). Doses evaluated ranged from 10 to 960 μg/kg/d for 24 h.
Trends in improvement in a number of hemodynamic parameters were observed during dosing and for several hours thereafter. Doses of relaxin in the range of 10 to 100 μg/kg/d appeared to have a more pronounced effect than higher doses on right atrial pressure, pulmonary artery pressure, PCWP, and NT-pro BNP, whereas the effects on cardiac index and SVR were more modest and appeared somewhat greater at higher doses. For example, the mean changes from baseline in PCWP at 30, 100, 480, and 960 μg/kg/d were −5.0 ± 2.6 mmHg, −3.5 ± 3.7 mmHg, 0.33 ± 2.6 mmHg, and −0.17 ± 2.7 mmHg, respectively. No consistent changes in heart rate or blood pressure were observed either during dosing or postdosing.
The different dose responses observed for the hemodynamic parameters may be explained by imbalances in baseline hemodynamic status of the dose groups or may be due to random variability in a small pilot study. However, certain biological effects of relaxin are known to follow a U-shaped dose-response curve [25–27]. Alternatively, counter-regulatory neurohormonal mechanisms may have been stimulated at higher relaxin doses, thus overpowering the salutary responses observed at the lower doses. An intriguing alternative explanation may be that doses in the lower range produce more venous vasodilation (decrease in PCWP without change in CO/CI), whereas higher doses may produce more arterial vasodilation. Additional well-controlled hemodynamic studies will be required to provide further insight into this pattern.
Based on these hemodynamic effects, relaxin was subsequently tested in the Pre-RELAX-AHF Phase II trial. This trial was designed to assess drug effects on multiple end points, including symptoms, signs, and short-term outcomes in patients with AHF. A total of 234 patients hospitalized with dyspnea due to AHF were randomly assigned to receive placebo or one of four active doses of relaxin (10, 30, 100, or 250 μg/kg/d) for 48 h. A clear vasodilatory effect of relaxin was seen in the greater reduction of blood pressure in relaxin-treated patients compared with those receiving placebo, despite greater use of diuretics and intravenous nitroglycerin in the placebo group [10••].
The effects of relaxin on blood pressure corresponded directly to the relief of dyspnea, which were rapid and clinically meaningful, consistent with effects seen with other vasodilators. However, unlike other vasodilators, the effects of relaxin were sustained to day 14 (Table 1). It is possible that the impact of relaxin on both vasodilation and natriuresis/diuresis (see section below) may have led to more complete resolution of congestion, thereby fostering a prolonged amelioration of symptoms. Patients treated with relaxin also demonstrated trends toward greater resolution of congestion, measured as improvements in edema and pulmonary rales, as well as a shorter length of index hospital stay, and improved morbidity and mortality. Greater decongestion in patients with AHF has previously been associated with improved prognosis during the first 2 to 6 months following discharge [28, 29]. In addition, the proportion of patients demonstrating inhospital worsening heart failure during the first 5 days (defined as an increase in symptoms requiring intervention) was lower in the groups receiving relaxin compared with those receiving placebo. Early recurrence or worsening of heart failure has been associated with poor intermediate-term prognosis and may reflect persistence of underlying pathophysiologic mechanisms in AHF, including neurohormonal, inflammatory, and ischemic mechanisms [30].
The dose in Pre-RELAX-AHF that was associated with the greatest benefit was 30 μg/kg/d; this dose is being tested further in the ongoing Phase III RELAX-AHF study.
Interestingly, in Pre-RELAX-AHF, a greater vasodilatory effect and corresponding improvement in dyspnea [31] were observed in patients presenting with higher baseline blood pressure. This is consistent with observations made in nonclinical studies, in which hemodynamic effects of relaxin were proportional to the extent of baseline abnormality [27]. Also, in a clinical trial of 239 patients with progressive systemic sclerosis (PSS), relaxin caused small but statistically significant reductions in blood pressure of approximately 5 mmHg, consistent with changes observed in pregnancy, during 6 months of continuous therapy [13••]. A greater degree of blood pressure reduction (15–20 mmHg) was observed in the subset of patients who were hypertensive at baseline compared with those who were normotensive.
Clinical Data on Renal Effects
A number of clinical trials have suggested that relaxin stimulates increases in renal blood flow (RBF), consistent with a large body of nonclinical research indicating that relaxin causes vasodilation of the renal vasculature. A study of relaxin administered to healthy volunteers directly measured its effects on RBF, glomerular filtration rate (GFR), and electrolyte clearance [32]. Eleven subjects received 5-hour infusions of relaxin that led to serum concentrations approximating those seen in pregnancy, and GFR and RBF were measured using inulin and para-aminohippurate, respectively.
Consistent with the effects of endogenously circulating relaxin in pregnancy, relaxin administration was associated with a significant 47% increase in RBF, with evidence of an increase within 30 min of relaxin infusion. No increase in GFR was measured. Relaxin-stimulated increases in clearance and urinary excretion of sodium were also measured, consistent with nonclinical models [33].
In the Pre-RELAX-AHF heart failure study, there were consistent trends toward increased weight loss in the relaxin treatment groups that were measured at multiple time points, including 24 and 48 h of dosing and at 5 days and at 14 days postdosing [10••]. Although statistical significance was seen only at some time points, the overall trend supports the hypothesis that relaxin may have had a natriuretic/diuretic effect in these heart failure patients.
Although the study examining relaxin’s renal pharmacology did not show that an increase in GFR accompanied the elevation in RBF [32], other clinical data do suggest that relaxin may be able to modulate GFR. In the study of relaxin in patients with systemic sclerosis, there was a statistically significant 10% to 15% increase in the predicted creatinine clearance associated with relaxin administration throughout the 6-month treatment period, suggesting an increase in GFR [13••, 34].
Similarly, in 16 patients with stable AHF, decreases in serum creatinine and BUN (blood urea nitrogen) were observed during the 24-hour relaxin infusion and for an additional 24 h thereafter [9••]. At higher doses of drug, however, 1 week after completion of dosing, creatinine and BUN levels had returned to levels that were higher than baseline. There were no clinical consequences associated with these observed laboratory value changes.
In the Pre-RELAX-AHF study, no consistent changes in serum creatinine were observed during the relaxin infusion in AHF patients [10••]. It is possible that other medications may have interfered with a relaxin effect or that a difference in the underlying renal biology in these patients affects the response to relaxin. At the highest dose of relaxin tested, postdosing trends toward increased creatinine levels were observed. There were no clinical correlations of these laboratory value changes.
Anti-inflammatory Effects and Prevention of Reperfusion Injury
Inflammation appears to play a pivotal role in the pathogenesis of AHF and its poor prognosis [3, 30]. Animal models have provided data that indicate that relaxin, whose receptors are localized in the heart [35–37], may have anti-inflammatory properties and protective effects against reperfusion injury. Relaxin inhibits histamine release by mast cells in rat and guinea pig models of inflammation [38]. It also inhibits the influx of neutrophils into injured organs [39] and decreases the release of tumor necrosis factor (TNF)-α in response to adjuvant [40] and the release of interleukin (IL)-1, IL-6, and TNF-α in response to endotoxin [41]. These anti-inflammatory effects may contribute to the protection against cardiac reperfusion injury seen with relaxin therapy in rodent [42, 43] and swine models [44, 45]. In a recent study in a swine model [45], relaxin administration upon reperfusion following 30 min of ischemia inhibited plasma histamine levels, cardiac histamine content, and cardiac mast cell degranulation. Attendant with these effects, relaxin inhibited oxidative cardiac tissue injury and the occurrence of severe ventricular arrhythmias, both consequences of reperfusion-induced inflammation. Because cardiac arrhythmias contribute to mortality following myocardial infarction, these findings are of potential significance.
Extracellular Matrix Remodeling Effects
Relaxin has demonstrated the ability to modify extracellular matrix deposition and turnover in multiple animal models. Relaxin, infused for as short a period as 2 to 3 days in rats, has been shown to increase global arterial compliance, coincident with an increase in cardiac output and decrease in SVR [46] and consistent with its role in pregnancy [27]. The increase in compliance studied in an ex vivo model showed a relaxin-mediated increase in compliance independent of smooth muscle-mediated changes in tone, suggesting that alterations in the extracellular matrix of the vessel wall contributed to increased compliance.
Systemic arterial compliance makes an important contribution to cardiac afterload, and thus, is a significant determinant of left ventricular work. Previous studies have suggested that arterial compliance may be reduced in patients with congestive heart failure. The ability of relaxin to increase arterial compliance through vasodilatory and antifibrotic effects may contribute to its beneficial effects in heart failure and warrant further investigation [47, 48].
Male relaxin-deficient mice demonstrate an age-related progression of cardiac fibrosis. Treatment for 2 weeks with relaxin of established cardiac fibrosis in these mice reduced collagen deposition [37]. Infusion for 14 days of relaxin reversed cardiac fibrosis in transgenic animals overexpressing β2 adrenergic receptors, which eventually develop heart failure [37]. In the spontaneously hypertensive rat model, relaxin therapy significantly decreased collagen content in the myocardium of the left ventricle, but not the unaffected chambers of the heart [49]. In addition, relaxin has shown similar antifibrotic effects in animal models of renal and pulmonary fibrosis [50–52].
Angiogenesis, Anti-ischemic Effects
Ischemic heart disease is a common underlying cause of heart failure. In fact, concurrent ischemia as evidenced by detectable levels of troponin identifies a group of patients with AHF who are at particularly high risk for morbidity and mortality [53]. In the Pre-RELAX-AHF study [10], a post-hoc exploratory analysis suggested that patients with detectable troponin at baseline derived the most benefit from relaxin therapy with respect to longer-term morbidity and mortality (Data on File, Corthera, Inc.). Relaxin’s angiogenic properties were demonstrated using a model of chronic myocardial infarction in rats [54]. Systemic infusion of relaxin potentiated the increase in basic fibroblast growth factor (bFGF) mRNA and protein at 7 and 21 days in the peri-infarct region being expressed by myocytes and fibroblasts. Similar infusions in sham-operated rats showed no change in bFGF or vascular endothelial growth factor expression in corresponding regions of the left ventricle or in the right ventricle. The increase in the number of thin-walled, collateral vessels lacking smooth muscle was correspondingly potentiated in the peri-infarct region by systemic relaxin administration. These proangiogenic and anti-ischemic effects, coupled with the anti-reperfusion injury effects described above, could potentially play a beneficial role in patients with AHF.
Conclusions
Relaxin is a naturally occurring peptide hormone first identified for its role in reproduction and now understood to play a central role in hemodynamic, renal, and other physiologic and pathologic processes. An appreciation of its hemodynamic and renal physiologic effects has led to its evaluation as a potential treatment of AHF with encouraging results. These results can be directly understood in the context of the known biology of relaxin, including favorable effects on multiple mechanisms ranging from central hemodynamics to kidney function, myocardial ischemia, and systemic inflammation (Table 2). Further evidence of therapeutic benefit, however, is still needed from large multicenter trials such as the ongoing RELAX-AHF, a phase III study of relaxin in patients hospitalized with AHF.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Cotter G, Metra M, Milo-Cotter O, et al.: Fluid overload in acute heart failure-re-distribution and other mechanisms beyond fluid accumulation. Eur J Heart Fail 2008, 10:165–169.
• Cotter G, Felker GM, Adams KF, et al.: The pathophysiology of acute heart failure-is it all about fluid accumulation? Am Heart J 2008, 155:9–18. This article provides a broader insight into the pathophysiology of AHF.
Colombo PC, Onat D, Sabbah HN: Acute heart failure as “acute endothelitis”-interaction of fluid overload and endothelial dysfunction. Eur J Heart Fail 2008, 10:170–175.
Hisaw FL: Experimental relaxation of the pubic ligament of the guinea pig. Proc Soc Exp Biol Med 1926, 23:661–663.
Fevold HL, Hisaw FL, Meyer RK: The relaxative hormone of the corpus luteum. Its purification and concentration. J Am Chem Soc 1930, 52:3340–3348.
•• Jeyabalan A, Shroff SG, Novak J, et al.: The vascular actions of relaxin. Adv Exp Med Biol 2007, 612:65–87. This article is an extensive review of the vascular biology and pharmacology of relaxin.
Schrier RW, Durr JA. Pregnancy: an overfill or underfill state. Am J Kidney Dis 1987, 9:284–289.
Baylis C: Relaxin may be the “elusive” renal vasodilatory agent of normal pregnancy. Am J Kidney Dis 1999, 34:1142–1145.
•• Dschietzig T, Teichman SL, Unemori E, et al.: Intravenous recombinant human relaxin in compensated heart failure: a safety, tolerability, and pharmacodynamic trial. J Card Fail 2009, 15:182–190. This phase 1 study provided cardiac hemodynamic proof-of-concept data for the vasodilatory properties of relaxin.
•• Teerlink JRT, Metra M, Felker GM, et al.: Relaxin for the treatment of patients with acute heart failure (Pre-RELAX-AHF). Lancet 2009, 373:1429–1439. This phase 2 study provided encouraging data that relaxin could relieve symptoms in patients with AHF and possibly improve prognosis.
Conrad KP, Novak J: Emerging role of relaxin in renal and cardiovascular function. Am J Physiol Regul Integr Comp Physiol 2004, 287:R250–R261.
Dschietzig T, Bartsch C, Baumann G, et al.: Relaxin-A pleiotropic hormone and its emerging role for experimental and clinical therapeutics. Pharmacol Therapeutics 2006, 112: 38–56.
•• Teichman SL, Unemori E, Dschietzig T, et al.: Relaxin, a pleiotropic vasodilator for the treatment of heart failure. Heart Fail Rev 2009, 14:321–329. This extensive review discusses the vascular biology, pharmacology, and clinical data of relaxin, with a focus on the potential to treat heart failure.
Samuel CS, Hewitson TD, Unemori EN, Tang MLK: Drugs of the future: the hormone relaxin. Cell Mol Life Sci 2007, 64:1539–1557.
Haynes WG, Webb DJ: Endothelin as a regulator of cardiovascular function in health and disease. J Hypertens 1998, 16:1081–1098.
Schneider MP, Boesen EI, Pollock DM: Contrasting actions of endothelin ETA and ETB receptors in cardiovascular disease. Ann Rev Pharmacol Toxicol 2007, 47:731–759.
McMurray JJ, Ray SG, Abdullah I, et al.: Plasma endothelin in chronic heart failure. Circulation 1992, 85:1374–1379.
Good JM, Nithoyannopoulos P, Ghatei MP, et al.: Elevated plasma endothelin concentrations in heart failure; an effect of angiotensin II? Eur Heart J 1994, 15:1634–1640.
Masson S, Latini R, Anand IS, et al.: The prognostic value of big endothelin-1 in more than 2,300 patients with heart failure enrolled in the Valsartan Heart Failure Trial (Val-HeFT). J Card Fail 2006, 12:375–380.
Asano K, Bohlmeyer TJ, Westcott JY, et al.: Altered expression of endothelin receptors in failing human left ventricles. J Molec Cell Cardiol 2002, 34:833–846.
Bogzil AH, Ashton N: Relaxin-induced changes in renal function and RXFP1 receptor expression in the female rat. Ann N Y Acad Sci 2009, 1160:313–316.
Dschietzig T, Richter C, Bartsch C, et al.: The pregnancy hormone relaxin is a player in human heart failure. Faseb J 2001, 15: 2187–2195.
Dschietzig T, Bartsch C, Richter C, et al.: Relaxin, a pregnancy hormone, is a functional endothelin-1 antagonist: attenuation of endothelin-1-mediated vasoconstriction by stimulation of endothelin type-B receptor expression via ERK-1/2 and nuclear factor-kappaB. Circ Res 2003, 92: 32–40.
Danielson LA, Sherwood OD, Conrad KP: Relaxin is a potent renal vasodilator in conscious rats. J Clin Invest 1999, 103:525–533.
Unemori EN, Amento EP: Relaxin modulates synthesis and secretion of procollagenase and collagen by human dermal fibroblasts. J Biol Chem 1990, 265:10681–10685.
Danielson LA, Conrad KP: Time course and dose response of relaxin-mediated renal vasodilation, hyperfiltration, and changes in plasma osmolality in conscious rats. J Appl Physiol 2003, 95:1509–1514.
Debrah DO, Conrad KP, Jeyabalan A, et al.: Relaxin increases cardiac output and reduces systemic arterial load in hypertensive rats. Hypertension 2005, 46:745–750.
Lucas C, Johnson W, Hamilton MA, et al.: Freedom from congestion predicts good survival despite previous class IV symptoms of heart failure. Amer Heart J 2000, 140:840–847.
Costanzo MR, Guglin ME, Saltzberg MT, et al.: Ultrafiltration versus intravenous diuretics for patients hospitalized for acute decompensated heart failure. J Amer Coll Cardiol 2007, 49:675–683.
Milo O, Cotter G, Kaluski E, et al.: Inflammatory and neurohormonal activation in cardiogenic pulmonary edema: implications on the pathogenesis and outcome of acute ischemic versus non-ischemic acute heart failure. Am J Cardiol 2003, 92:222–226.
Teerlink JR, Metra M, Felker GM, et al.: Vasodilators in acute heart failure (AHF): does blood pressure matter? Results from Pre-Relax-AHF. J Cardiac Failure 2009, 15:S74.
Smith MC, Danielson LA, Conrad KP, et al.: Influence of recombinant human relaxin on renal hemodynamics in healthy volunteers. J Am Soc Nephrol 2006, 17:3192–3197.
Bogzil AH, Eardley R, Ashton N: Relaxin-induced changes in renal sodium excretion in the anesthetized male rat. Am J Physiol Regulatory Integrative Comp Physiol 2005, 288:R322–R328.
Erikson MS, Unemori E: Relaxin clinical trials in systemic sclerosis. In Relaxin 2000, Proceedings of the Third International Conference on Relaxin and Related Peptides. Edited by Tragear GW, Ivell R, Bathgate RA, Wade JD. Broome, Australia: Kluwe Academic Publishers; 2000:373–381.
Hsu SY, Nakabayashi K, Nishi S, et al.: Activation of orphan receptors by the hormone relaxin. Science 2002, 295:671–674.
Kompa AR, Samuel CS, Summers RJ: Inotropic responses to human gene 2 (B29) relaxin in a rat model of myocardial infarction (MI): effect of pertussis toxin. Br J Pharmacol 2002, 137:710–718.
Samuel CS, Unemori EN, Mookerjee I, et al.: Relaxin modulates cardiac fibroblast proliferation, differentiation and collage production and reverses cardiac fibrosis in vivo. Endocrinology 2004, 145:4125–4133.
Bani D, Ballati L, Masini E, et al.: Relaxin counteracts asthma-like reaction induced by inhaled antigen in sensitized guinea pigs. Endocrinology 1997, 138:1909–1915.
Masini E, Nistri S, Vannacci A, et al.: Relaxin inhibits the activation of human neutrophils: involvement of the nitric oxide pathway. Endocrinology 2004, 145:1106–1112.
Santora K, Rasa C, Visco D, et al.: Antiarthritic effects of relaxin, in combination with estrogen, in rat adjuvant-induced arthritis. J Pharmacol Exp Ther 2007, 322:887–893.
Dschietzig T, Bartsch C, Stangl V, et al.: Identification of the pregnancy hormone relaxin as a glucocorticoid receptor agonist. FASEB J 2004, 18:1536–1538.
Masini E, Bani D, Bello MG, et al.: Relaxin counteracts myocardial damage induced by ischemia-reperfusion in isolated guinea pig hearts: evidence for an involvement of nitric oxide. Endocrinology 1997, 138:4713–4720.
Bani D, Masini E, Bello MG, et al.: Relaxin protects against myocardial injury caused by ischemia and reperfusion in rat heart. Amer J Pathol 1998, 152:1367–1376.
Perna AM, Masini E, Nistri S: Human recombinant relaxin reduces heart injury and improves ventricular performance in a swine model of acute myocardial infarction. Ann NY Acad Sci 2005, 1041:431–433.
Nistri S, Cinci L, Perna AM, et al.: Mast cell inhibition and reduced ventricular arrhythmias in a swine model of acute myocardial infarction upon therapeutic administration of relaxin. Inflamm Res 2008, 57(Suppl 1):S7–S8.
Conrad KP, Debrah DO, Novak J, et al.: Relaxin modifies systemic arterial resistance and compliance in conscious, nonpregnant rats. Endocrinology 2004, 145:3289–3296.
Samuel CS, Du XJ, Bathgate RA, Summers RJ: Relaxin the stiffened heart and arteries: the therapeutic potential for relaxin in the treatment of cardiovascular disease. Pharmacol Ther 2006, 112:529–552.
van der Westhuizen ET, Halls ML, Samuel CS, et al.: Relaxin family peptide receptors-from orphans to therapeutic targets. Drug Discov Today 2008, 13:640–651.
Lekgabe ED, Kiriazis H, Zhao C, et al.: Relaxin reverses cardiac and renal fibrosis in spontaneous hypertensive rats. Hypertension 2005, 46:412–418.
Lekgabe ED, Su Y, McGuane JT, et al.: Adenovirus-mediated delivery of relaxin reverses cardiac fibrosis in mice with fibrotic cardiomyopathy. J Mol Cell Cardiol 2006, 41:739.
Danielson LA, Welford A, Harris A: Relaxin improves renal function and histology in aging Munich-Wistar rats. J Am Soc Nephrol 2006, 17:1325–1333.
Tozzi CA, Poiani GJ, McHugh NA, et al.: Recombinant human relaxin reduces hypoxic pulmonary hypertension in the rat. Pulm Pharmacol Ther 2005, 18:346–353.
Peacock WF, De Marco T, Fonarow GC, et al.: Cardiac troponin and outcome in acute heart failure. N Engl J Med 2008, 358:2117–2126.
Lewis M, Deshpande U, Guzman L, et al.: Systemic relaxin administration stimulates angiogenic cytokine expression and vessel formation in a rat myocardial infarct model. In Relaxin 2000, Proceedings of the Third International Conference on Relaxin and Related Peptides. Edited by Tragear GW, Ivell R, Bathgate RA, Wade JD. Broome, Australia: Kluwe Academic Publishers; 2000:373–381.
Disclosure
Dr. Sam L. Teichman and Dr. Elaine Unemori are employees of Corthera, Inc. Dr. Gad Cotter is an employee of Momentum Research. In 2009, Dr. Marco Metra received honoraria and travel reimbursements from Corthera, Inc. No other potential conflicts of interest relevant to this article were reported.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Teichman, S.L., Unemori, E., Teerlink, J.R. et al. Relaxin: Review of Biology and Potential Role in Treating Heart Failure. Curr Heart Fail Rep 7, 75–82 (2010). https://doi.org/10.1007/s11897-010-0010-z
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11897-010-0010-z