Article Text

Abstract
Background Circulatory failure after out-of-hospital cardiac arrest (OHCA) as part of the postcardiac arrest syndrome (PCAS) is believed to be caused by an initial myocardial depression that later subsides into a superimposed vasodilatation. However, the relative contribution of myocardial dysfunction and systemic inflammation has not been established. Our objective was to describe the macrocirculatory and microcirculatory failure in PCAS in more detail.
Methods We included 42 comatose patients after OHCA where circulatory variables were invasively monitored from admission until day 5. We measured the development in cardiac power output (CPO), stroke work (SW), aortic elastance, microcirculatory metabolism, inflammatory and cardiac biomarkers and need for vasoactive medications. We used survival analysis and Cox regression to assess time to norepinephrine discontinuation and negative fluid balance, stratified by inflammatory and cardiac biomarkers.
Results CPO, SW and oxygen delivery increased during the first 48 hours. Although the estimated afterload fell, the blood pressure was kept above 65 mmHg with a diminishing need for norepinephrine, indicating a gradually re-established macrocirculatory homoeostasis. Time to norepinephrine discontinuation was longer for patients with higher pro-brain natriuretic peptide concentration (HR 0.45, 95% CI 0.21 to 0.96), while inflammatory biomarkers and other cardiac biomarkers did not predict the duration of vasoactive pressure support. Markers of microcirculatory distress, such as lactate and venous-to-arterial carbon dioxide difference, were normalised within 24 hours.
Conclusion The circulatory failure was initially characterised by reduced CPO and SW, however, microcirculatory and macrocirculatory homoeostasis was restored within 48 hours. We found that biomarkers indicating acute heart failure, and not inflammation, predicted longer circulatory support with norepinephrine. Taken together, this indicates an early and resolving, rather than a late and emerging vasodilatation.
Trial registration NCT02648061.
- heart arrest
- epidemiology
- heart failure
- biomarkers
Data availability statement
Data are available upon reasonable request. The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Key messages
What is already known about this subject?
The postcardiac arrest syndrome compromise brain injury; myocardial dysfunction; and systemic inflammation response. In addition, patient comorbidities and the pathological process that caused the cardiac arrest often complicate the clinical course. The circulatory failure is believed to be caused by an initial myocardial depression that later subsides into a superimposed vasodilatation. Still, the relative contribution of myocardial dysfunction and systemic inflammation has not been established.
What does this study add?
This prospective cohort study provides insight into the macrocirculatory and microcirculatory failure after cardiac arrest, with focus on the development of vasopressor need, energy and oxygen delivery, vascular resistance, oxygen consumption, lactate level and veno-arterial pCO2 gap. In addition, the study explored the relative contribution of inflammation and cardiac dysfunction on circulatory failure after cardiac arrest.
How might this impact on clinical practice?
In the literature the inflammatory vasodilatation after cardiac arrest is described as a later occurring phenomena that prevents discontinuation of vasopressors. However, in this study the vasodilatation was clinically early and resolving. Furthermore, biomarkers indicating acute heart failure, and not inflammation, predicted longer circulatory support with norepinephrine. These findings might influence the understanding of the underlying pathophysiology and act as a reminder of careful interpretation of vascular resistance measurements.
Introduction
Circulatory failure is frequently observed after out-of-hospital cardiac arrest (OHCA) as part of the postcardiac arrest syndrome (PCAS).1 The current understanding is that the circulatory failure in PCAS is mainly caused by myocardial depression followed by superimposed inflammatory vasodilatation.2–5 However, the interpretation of previous studies is challenging for several reasons. First, few studies use advanced haemodynamic measurements.6 7 Second, some studies report systemic vascular resistance (SVR) without complete information about each component of this equation.4 Third, there is commonly a lack of detailed information on vasoactive medications and fluids used to achieve the haemodynamic results.8 Fourth, therapeutic hypothermia has cardiovascular effects, such as bradycardia and vasoconstriction.9 Fifth, the heterogeneous nature of patients’ circulatory failure complicates the picture and there is reason to believe that there exist several different ‘circulatory phenotypes’ in patients with PCAS.10–12 Finally, the aetiology of circulatory failure is mixed, and the relative contribution of myocardial dysfunction and systemic inflammation after global ischemia and reperfusion is yet to be established.13 Thus, a more detailed description of circulatory disturbances after OHCA is warranted.
The total energy delivery from the heart to the circulation is measured per beat by stroke work (SW) or per minute by cardiac power output (CPO). Either way, the energy is in the form of a pressurised volume, which, dependent on the resistance to ejection, is distributed predominantly as flow or pressure. This balance between the heart’s contractility and its afterload is described as ventriculo-arterial coupling.14 CPO is the product of mean arterial pressure (MAP) and cardiac output (CO) and have been reported to correlate more strongly with mortality in cardiogenic shock than the variables from which it is derived.15–18 However, like all CO-derived quantities, both CPO and SVR depend on heart rate. In comparison, SW and aortic elastance (Ea) is not affected by heart rate and reflects better the heart’s beat-to-beat contractile function and afterload,19 especially in patients with possible hypothermia-induced bradycardia.
The aim of this study was to describe the circulatory failure in PCAS, with focus on the development of vasopressor need, energy and oxygen delivery, vascular resistance, oxygen consumption, lactate level and veno-arterial pCO2 gap. In addition, the study explored the relative contribution of inflammation and cardiac dysfunction on circulatory failure in PCAS.
Methods
Study design
This was a preplanned analysis of a previously published single-centre, prospective, observational cohort study of 50 patients admitted to hospital with return of spontaneous circulation (ROSC) after OHCA.12 20 Patients were included between January 2016 and November 2017. Only patients who were comatose at arrival were included in the current analysis.
Setting and eligibility
St. Olav’s University Hospital is a 938-bed tertiary hospital in Trondheim, Norway, serving a population of approximately 700 000. Comatose adults admitted to the intensive care unit (ICU) with ROSC after OHCA were included in this study. Exclusion criteria were age <18 years, pregnancy, assumed septic or anaphylactic aetiology of cardiac arrest, transferal from another hospitals, decision to limit life-sustaining therapy on arrival, acute cardiothoracic surgery, intervention with extracorporeal membranous oxygenation (ECMO) or a ventricular assist device (VAD) before arrival at the ICU.
Study procedure
We followed the patients from time of ICU admission and the following 5 days, or until the patient died, ECMO, VAD or acute cardiothoracic surgery was necessary, life-prolonging therapy was limited, or if the patient was transferred to a general ward or another hospital during the study period. Day 0 had variable length depending on the time of inclusion, whereas day 1 started the following morning at 06:00.
All patients without contraindications received a pulmonary artery catheter (PAC) (Swan-Ganz CCombo, Edwards Lifesciences, USA) for continuous central haemodynamic measurements. Bleeding diathesis and implanted pacemaker were the main contraindications. If technical difficulties were experienced during catheter placement, the procedure was aborted. We calibrated the SvO2 sensor two times a day.
We retrieved systolic and MAP, fluid administration, peripheral oxygen saturation (SpO2), temperature, lactate concentration, arterial and venous partial pressure of carbon dioxide (CO2), haemoglobin concentration (Hgb), heart rate and norepinephrine dosage from the electronic critical care management system (Picis CareSuite, Optum, USA). We retrieved CO, mean pulmonary artery pressure, stroke volume (SV) and mixed venous saturation (SvO2) from patients with a PAC. Continuous data were sampled in 1 min resolution, and presented in 1-hour intervals. We obtained time from emergency call to advanced cardiovascular life support (ACLS), ROSC and ICU admission, initial cardiac rhythm, number of defibrillations, aetiology, and whether the arrest were witnessed and cardiopulmonary resuscitation commenced, all according to the Utstein cardiac arrest template,21 Charlson Comorbidity Index22 and clinical information on assessment and treatment from the prehospital and hospital records. Echocardiography was performed in all patients within 24 hours after admission by experienced cardiologists.
Simplified Acute Physiology Score 2 were registered 24 hours after admission,23 and survival was registered after 30 days.
Haemodynamic calculations
CPO is measured in Watt (W) and calculated by CO (L/min)×MAP (mmHg) divided by 451. The normal resting CPO is ~1 W where <0.54 W is indicative of haemodynamic compromise.24 25 SW is measured in Joule (J) and calculated by CPO×60 divided by heart rate. Clinically, Ea can be approximated non-invasively by systolic blood pressure (mmHg)×0.9 divided by SV (mL), with a normal range 1.4–3 mmHg/mL.26 27 Oxygen delivery (DO2) and consumption (VO2) was calculated by 0.134×Hgb (g/dL)×SpO2 (%)×CO (L/min) and 0.134×Hgb (g/dL)×(SpO2–SvO2 (%))×CO (L/min), respectively.28
Blood samples
Interleukin (IL)-6, syndecan-1, pro-brain natriuretic peptide (BNP), troponin T and C reactive protein (CRP) concentrations were measured at inclusion and every morning during the study period. A detailed description of the blood sample handling is given in online supplemental text 1. Every 6 hours, we obtained an arterial blood gas sample, and two times a day a mixed venous blood gas sample, from which venous-to-arterial carbon dioxide difference (P(v–a)CO2, kPa) was calculated.
Supplemental material
Postcardiac arrest care and cardiovascular support
The patients were cooled to 36°C for 24 hours, with avoidance of hyperthermia (>37.5°C) for an additional 48 hours afterwards. Patients with suspected myocardial ischaemic aetiology of the cardiac arrest received coronary angiography and percutaneous revascularisation.
In the presence of hypotension (<65 mmHg) and clinical signs of tissue hypo-perfusion (such as cold clammy skin, prolonged capillary refill time, reduced urine output, increasing lactate and/or decreasing base excess), the circulation was optimised by fluid and vasopressor administration, based on the department’s guidelines on circulatory support. A detailed description of the postcardiac arrest care in this study has been published previously.20
Statistical analysis
PCAS has been described as a ‘sepsis-like syndrome’,2 and we hypothesise a prolonged vasopressor need, associated with high inflammatory biomarker concentrations, and a lack of macrocirculatory and microcirculatory coherence.
The mean circulatory development over time was graphically presented, with origin set to time of emergency call, and smoothed with 3 hours moving average to better visualise the trend. The differences between mean level of CPO, SW, Ea, SV, MAP and norepinephrine dosage at four and 48 hours after ICU admission were tested by Welch’s unequal variance t-test.
We used survival analysis to explore the time to discontinuation of norepinephrine and daily negative fluid balance as a marker of circulatory stabilisation. Dobutamine was sporadic used in four patients and therefore not analysed. As the main aetiologies of circulatory shock in PCAS are inflammatory and/or cardiogenic, we compared high versus low levels of IL-6, syndecan-1, CRP, pro-BNP and troponin T, where time of emergency call served as origin for timing of biomarker measurement. Based on available blood samples, we stratified the population by the median of the area under the curve (AUC) for the biomarker measurements the first 52 hours after the emergency call.29 Further, we used Cox regression to estimate the HR with 95% CI. Finally, we explored, with logistic regression, the demographic characteristics associated with the high biomarker group that significantly affected the duration of circulatory support.
Data were extracted with the software Matlab (Mathworks, USA), while the statistical analyses were performed with Stata V.16.0 (StataCorp). Obviously erroneous measurements (due to technical errors) were replaced by the mean value of the previous value and the value next minute.
Sample size
This was a descriptive study and no formal sample size calculation was performed.30
Results
Study population and outcomes
Sixty-five consecutive patients (eight missing) were assessed for eligibility. Of these, eight patients were excluded due to not being comatose, seven patients due to immediate withdrawal of life-support, two had septic aetiology, two patients not in need of ICU admission, three patients received VAD or ECMO and one patient underwent immediate surgery. Thus, 42 patients were included in this analysis. The mean length of day 0 was 12 hours (SD 5 hours) from ICU admission. Demographic and circulatory data are presented in table 1, and corresponding test results for patients with high versus low biomarker levels are presented in online supplemental table 1.
Supplemental material
Demography for all patients
Thirty-four of 42 patients received bystander cardiopulmonary resuscitation and 31 had shockable initial rhythm (table 1). From the time of the emergency call, the median time to ACLS was 10 min (first to third quartile (Q1–Q3) 5–15, min–max 0–36). ROSC was achieved after a median of 26 min (Q1–Q3 19–35, min–max 4–80), and median time to ICU admission was 133 min (Q1–Q3 102–168, min–max 26–263). Thirty patients (71 %) received a PAC. Within 30 days 15 of 42 patients had been declared dead.
Macrocirculation and circulatory support
Norepinephrine doses were gradually reduced, and after 48 hours, the mean dose of norepinephrine was halved, while the MAP had been maintained above 65 mmHg. Mean fluid balance was negative by day 4. Mean SV, SW and CPO increased from 50 mL, 0.5 J and 0.6 W on ICU admission to stable levels at approximately 80 mL, 0.8 J and 1 W, respectively, at 48 hours (figure 1A,B). Simultaneously, Ea was reduced approximately from 2.0 to 1.4 mmHg/mL (figure 1C). Additional macrocirculatory variables and more detailed echocardiographic results are presented in online supplemental figure 1 and table 2.
Supplemental material

Macrocirculatory variables and circulatory support in 42 patients admitted to ICU after OHCA. (A) Mean cardiac power output with 95% CIs. (B) Mean stroke work with 95% CIs. (C) Mean arterial elastance with 95% CIs. (D) Mean arterial and pulmonary blood pressure with 95% CIs. (E) Mean dosage of norepinephrine with 95% CIs. (F) Mean fluid balance during study period. The mean level of CPO, SW, Ea, norepinephrine dosage at 4 and 48 hours after admission were significantly different (p<0.05). The difference in MAP was not significant (p=0.45). The graphs were smoothed with a 3 hours moving average. Graphs A, B, C and D were based on the 30 patients with PAC. CPO, cardiac power output; Ea, aortic elastance; ICU, intensive care unit; MAP, mean arterial pressure; MPAP, mean pulmonary arterial pressure; OHCA, out-of-hospital cardiac arrest; SW, stroke work.
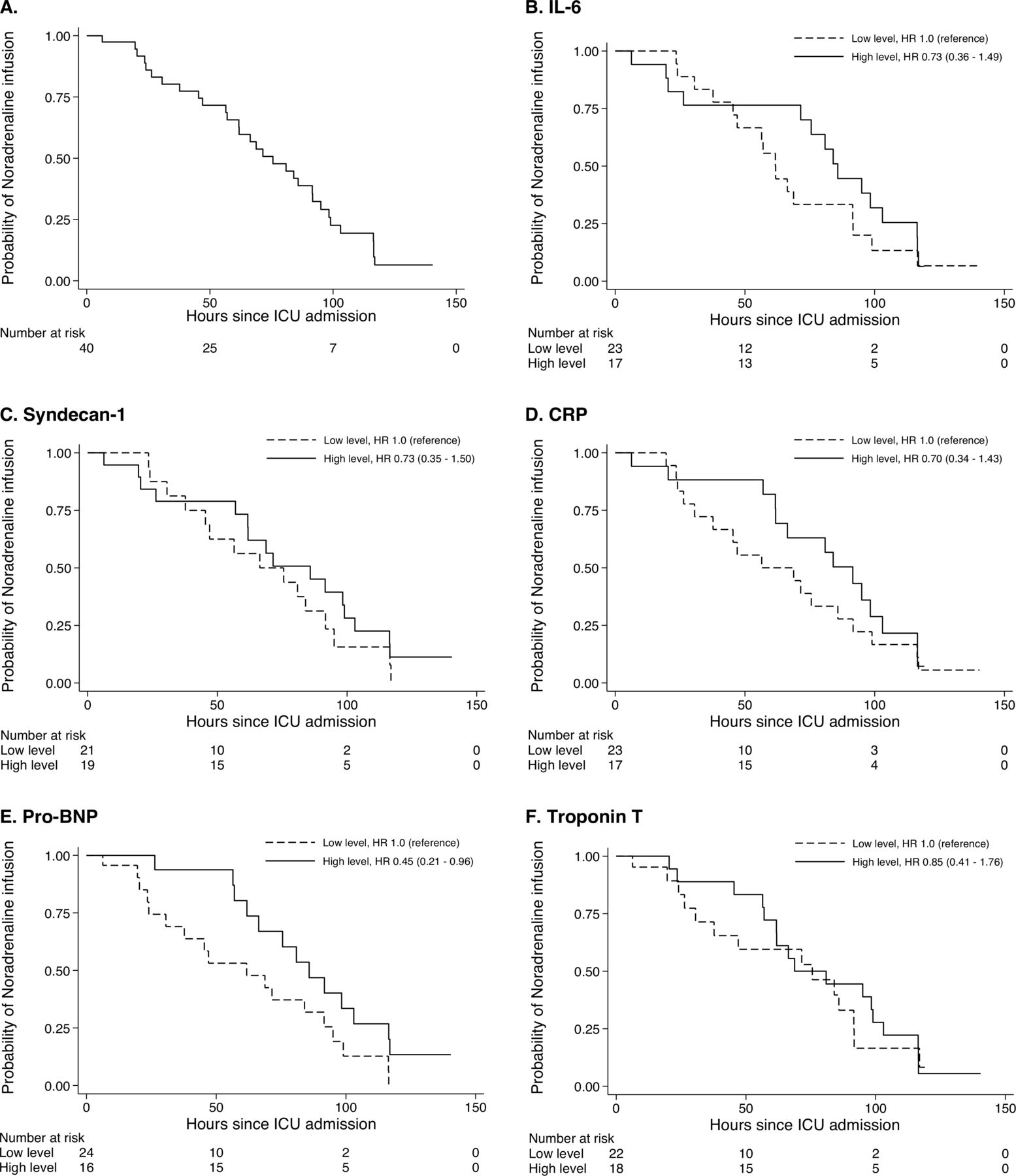
Kaplan-Meier estimates of the probability of on-going norepinephrine infusion and positive fluid balance are presented in figure 2 and online supplemental figure 2. The median time to discontinuation of norepinephrine was 75 hours for on-going norepinephrine infusion and the fourth morning for negative fluid balance. There was a significant difference in time to norepinephrine discontinuation between low versus high pro-BNP groups (HR 0.45, 95% CI 0.21 to 0.96), but not between the other groups. We found no significant difference between groups regarding time to negative fluid balance (online supplemental figure 2).
Supplemental material

Probability of norepinephrine infusion over time in 42 patients admitted to ICU after OHCA. Kaplan-Meier estimates of the probability of norepinephrine infusion for all patients (A) and stratified by biomarker level (B–F). The HR with 95% CIs was estimated by Cox regression. BNP, brain natriuretic peptide; CRP, C reactive protein; ICU, intensive care unit; IL, interleukin; OHCA, out-of-hospital cardiac arrest.
We found no significant association between patients in the high pro-BNP group and demographic variables (table 2).
Logistic regression analysis of association between high pro-brain natriuretic peptide and demographic variables
Oxygen transport and metabolism
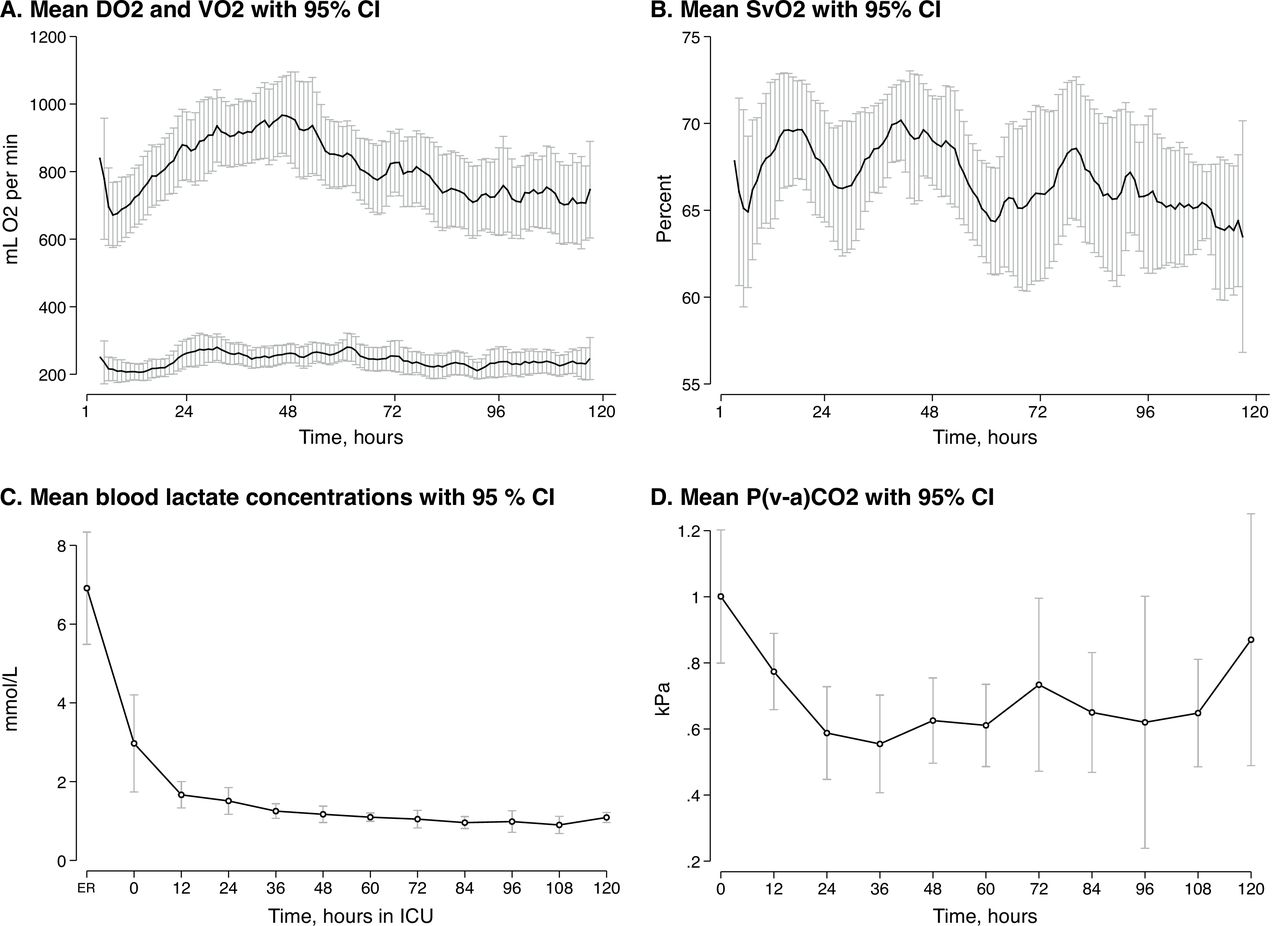
Mean VO2 was stable at approximately 200 mL/min, whereas DO2 increased from 700 to its peak 950 mL/min during the first 48 hours. Mean SvO2 was stable at approximately 68%. Mean lactate and P(v−a)CO2 levels were normalised within 24 hours (figure 3).
{kind=link}
{kind=link}
{kind=link}
Oxygen transport and metabolic distress variables in 42 patients admitted to ICU after OHCA. (A) Mean global oxygen delivery and consumption with 95% CIs. (B) Mean mixed venous oxygen saturation with 95% CIs. (C) Mean blood lactate concentrations with 95% CIs. (D) Mean venous-to-arterial carbon dioxide difference with 95% CIs. Graph A and B were smoothed with a 3 hours moving average. Graphs A, B and D were based on the 30 patients with PAC. DO2, oxygen delivery; ER, emergency room; ICU, intensive care unit; OHCA, out-of-hospital cardiac arrest; P(v−a)CO2, venous-to-arterial carbon dioxide difference; SvO2, mixed venous oxygen saturation; VO2, oxygen consumption.
Discussion
In this study, we found an overall significant increase in CPO, SW and SV during the first 48 hours in patients after OHCA. A stable MAP above 65 mmHg was achieved with initial norepinephrine and fluid infusions that could be reduced over time, even if Ea fell, due to gradually reestablishment of circulatory homoeostasis. We found a significant longer time to norepinephrine discontinuation in patients with high pro-BNP concentration.
The circulatory failure after OHCA has been described as first a reversible myocardial failure that improves over 24 hours, followed by an inflammatory vasodilatation necessitating continued vasopressor support and extra fluid infusion.3 In acute heart failure most of the cardiac reserves can be mobilised in order to sustain oxygen delivery, and the initial low CPO may indicate exhausted, or nearly exhausted, cardiac reserves due to post-arrest stunning that resolved over the next 48 hours.15 After 48 hours both the SW and CPO curves flatten, which could indicate a restored heart function. Furthermore, the similarity between the SW and CPO curves indicates that the increase in CPO was not due to an increasing heart rate. Heart rate is temperature dependent but lowering the body temperature to 36°C seemed to have a negligible bradycardic effect. In compliance with the current guidelines MAP were kept above 65 mmHg.1 13 This was achieved with norepinephrine infusion, which is the preferred vasoactive agent in PCAS.1 31 32 However, even if calculated Ea fell as a consequence of rising SV and stable blood pressure, this did not lead to an increase in norepinephrine nor fluid infusions. A similar trend in SVR and vasopressor doses has been shown previously.8 12 This should serve as a reminder that calculated resistance is not an exact measure of vascular tone, nor necessarily a clinical relevant vasoplegia. Thus, we propose that the inflammatory vasodilatation after OHCA is early and resolving, rather than late and emerging.
Microcirculatory failure is central to the circulatory pathogeneses in sepsis.33 34 It is believed that systemic inflammation-reperfusion injury after OHCA induces a similar circulatory failure.2 Two small studies using sidestream dark-field (SDF) imaging found reduced microcirculatory flow that improved over the 24 hours after OHCA.35 36 In presence of an adequate DO2, an increased P(v–a)CO2 difference is a sign of microcirculatory derangement, and in sepsis shown to correlate with SDF-evaluated microcirculatory flow alterations.37 Lactataemia in circulatory shock is multifactorial, but during shock commonly regarded as catecholamine-induced hyper-metabolism rather than sign of hypo-perfusion.38 Regardless of aetiology, lack of lactate clearance is a marker of sustained stressed metabolism, and associated with mortality in critically ill patients.39 In our study, both lactate and P(v–a)CO2 difference were normalised within 24 hours indicating improved microcirculation.
Taken together, improving macrocirculation and microcirculation with reduced need for circulatory support, and no difference between groups with high versus low inflammation (IL-6 and syndecan-1), it seems that PCAS lacks the self-sustaining inflammation seen in septic shock. In contrast, this study shows that the degree of initial heart failure (expressed by elevated pro-BNP) and not initial cardiac injury (elevated troponin), nor initial inflammation (elevated IL-6, syndecan-1 and CRP), was indicative for longer need of norepinephrine. This difference could not be explained by excess mortality in any of the groups.
Strengths and limitations
Patients in this study were treated at one centre adhering to one protocol, which renders the use of fluid therapy and vasopressors comparable. Furthermore, we measured and assessed the circulatory outcome and interventions in high resolution. However, a single-centre study limits the possible size of the study population, with the risk of making a type II error where significant relationships were missed. Both size and a single centre design reduced the generalisability of the results, however, from a clinical standpoint, the result of this study seemed reasonable. We stratified the groups based on the integral of the biomarker measurements the first 52 hours after the emergency call; acknowledging that a different time span could have produced a different result. Still, IL-6 and syndecan-1 concentrations peak early and decline rapidly after OHCA, thus the majority of the integral was covered during the first 48 hours.40 41 Troponin T and pro-BNP showed a similar pattern (online supplemental figure 3). Nonetheless, dichotomising the population based on AUC, rather than a single blood sample, is considered a more robust approach.29 Finally, sedation affects the need for vasopressors, and we did not examine the impact of late extubation due to respiratory or neurological conditions.42
Supplemental material
Conclusion
In this observational cohort study of 42 patients admitted to ICU after OHCA, the circulatory failure was initially characterised by reduced CPO and SW, however microcirculatory and macrocirculatory homoeostasis was restored within 48 hours. We found that biomarkers indicating acute heart failure, and not inflammation, predicted longer circulatory support with norepinephrine. Taken together, this indicates an early and resolving, rather than a late and emerging vasodilatation.
Supplemental material
Data availability statement
Data are available upon reasonable request. The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.
Ethics statements
Patient consent for publication
Ethics approval
This study involves human participants and was approved by Regional Committee for Medical and Health Research Ethics, Central Norway Health Region ID: REK Midt, No. 2015/1807 Comatose patients after cardiac arrest.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors HL, DB, NKS and PK included patients, initiated treatment and placed all pulmonary artery catheters in accordance with the study protocol. HL, DB, NKS, PK, TRS and KB supervised the study and patient care daily. HL, ML and TN contributed extensively to the statistical analysis. All authors contributed to interpreting the data and writing the manuscript. All authors have read and approved the final manuscript. HL is the guarantor for the overall content.
Funding This work is funded by a research grant to Dr Langeland from the Norwegian University of Science and Technology and St. Olav’s University Hospital (Samarbeidsorganet HMN NTNU).
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.