Article Text

Abstract
Background Myocardial infarction (MI) is associated with mental health disorders, in which neuroinflammation and cerebral microvascular dysfunction may play a role. Previously, we have shown that the proinflammatory factors Nε-(carboxymethyl)lysine (CML) and NADPH oxidase 2 (NOX2) are increased in the human infarcted heart microvasculature. The aim of this study was to analyse the presence of CML and NOX2 in the cerebral microvasculature of patients with MI.
Methods Brain tissue was obtained at autopsy from 24 patients with MI and nine control patients. According to their infarct age, patients with MI were divided into three groups: 3–6 hours old (phase I), 6 hours–5 days old (phase II) and 5–14 days old (phase III). CML and NOX2 in the microvasculature were studied through immunohistochemical analysis.
Results We observed a 2.5-fold increase in cerebral microvascular CML in patients with phase II and phase III MI (phase II: 21.39±7.91, p=0.004; phase III: 24.21±10.37, p=0.0007) compared with non-MI controls (8.55±2.98). NOX2 was increased in microvessels in patients with phase II MI (p=0.002) and phase III MI (p=0.04) compared with controls. No correlation was found between CML and NOX2 (r=0.58, p=0.13).
Conclusions MI coincides with an increased presence of CML and NOX2 in the brain microvasculature. These data point to proinflammatory alterations in the brain microvasculature that may underlie MI-associated mental health disorders.
- myocardial infarction
- inflammation
- biomarkers
Data availability statement
All data relevant to the study are included in the article or uploaded as supplementary information.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Key questions
What is already known about this subject?
Myocardial infarction (MI) can induce cerebral disease and cognitive decline. Recently, an increase in neuroinflammation, indicated by activated macrophages and microglia, has been shown in the brains of patients with MI, suggesting that neuroinflammation may play a role in MI-induced mental health disorders.
What does this study add?
In this study, we show increased levels of Nε-(carboxymethyl)lysine and NADPH oxidase 2 in the microvasculature of the brain in time after MI in patients. These results indicate the induction of cerebral microvascular dysfunction and highlight a possible role for the brain microvasculature in MI-induced brain pathology.
How might this impact on clinical practice?
These results increase our knowledge of MI-related cognitive impairment and aids the development of novel treatments.
Introduction
Myocardial infarction (MI) has been associated with cerebral diseases such as depression and dementia.1–4 Cerebral small vessel disease (CSVD), a generic term that covers a variety of cerebral microvascular abnormalities, including increased vessel permeability and inflammation, is a known risk factor for cognitive impairment.5 6 Importantly, CSVD was found to be increased both in clinically recognised and unrecognised studies of patients with MI.4 7 8 Recent studies have shown decreased cerebral blood flow after MI.9 In addition, positron emission tomography imaging of mice and patients after MI revealed increased microglial activity in the brain, indicative of increased neuroinflammation.10 Neuroinflammation, characterised by leucocyte activation and abnormal cytokine production, cerebral endothelial dysfunction and increased blood–brain barrier permeability are increasingly recognised as key components of cognitive impairment.10–14
MI can result in accumulation of advanced glycation end (AGE) products, specifically Nε-(carboxymethyl)lysine (CML) in the intramyocardial vasculature.15 16 CML accumulation is enhanced by inflammation and oxidative stress. CML, in turn, is positively involved in the induction of inflammation and reactive oxygen species (ROS) production.17 18 Furthermore, the ROS producer NADPH oxidase 2 (NOX2) is upregulated in the intramyocardial vasculature subsequent to MI, and it is known that CML and NOX2 can stimulate one another.19–21 It has been shown that CML accumulates in the cerebral microvasculature during ageing of atherosclerotic mice.22 Moreover, NOX2 was elevated in the cerebral arteries of mice after an ischaemic stroke.23 In addition, both accumulated CML and increased NOX2 activity in the cerebral vasculature are associated with cerebrovascular disease and cognitive impairment, although this was not studied in patients with MI.24 25
Since the underlying mechanisms that associate MI with cognitive impairment remain unclear, we have now studied whether MI in humans coincides with accumulation of CML and NOX2 in the microvasculature of the brain.
Methods
Patients
Brain tissue from the cerebrum was obtained during autopsy from 24 patients who died after MI and 9 patients who died without pathological evidence of heart or brain disease that were used as a control group (table 1). Brain tissue was consistently sampled from the same brain area in the cerebrum if there were no anomalies. Based on histological examination of the heart, the patients with MI were divided into three groups according to the age of the MI: 3–6 hours old (lactate dehydrogenase loss of cardiomyocytes but without infiltration of neutrophilic granulocytes, phase I); between 6 hours and 5 days old (infiltration of neutrophilic granulocytes in the heart, phase II); between 5 and 14 days old (granulation tissue formation in the heart, phase III).26 After excision, the brain tissue was immediately fixed in formalin and subsequently embedded in paraffin for immunohistochemical (IH) analysis.
Clinical characteristics of included patients
The age distribution of patients with phase I, phase II and phase III MIs and non-MI controls was compared with a one-way analysis of variance (ANOVA), displayed as mean±SD. The distribution of sex, diabetes mellitus (DM) and prior incidence of MI in non-MI controls and patients with MI, comprising the three phases of MI, was analysed with a χ2 test. A p value of <0.05 was considered statistically significant.
Immunohistochemistry
The paraffin-embedded brain tissue was cut into 4 µm thick sections and mounted onto microscope slides. The slides were first deparaffinised in xylene for 10 min, then dehydrated in 100% ethanol for 10 min and subsequently incubated in methanol containing 0.3% H2O2 for 30 min to block endogenous peroxidases. For antigen retrieval, the slides were incubated in 0.1% pepsin buffer for 30 min (for CML staining) or boiled in Tris/EDTA buffer (pH 9.0) for 15 min followed by cooling down for 15 min (for NOX2 staining). All sera and antibodies were diluted in normal antibody diluent solution (ImmunoLogic), and incubations were at room temperature unless otherwise specified. The tissue was blocked with either 2% (w/v) bovine serum albumin for CML staining, or with normal rabbit serum (NRS; 1:10 dilution, Dako, Glostrup, Denmark) for NOX2 staining. Subsequently, the slides were incubated with either mouse anti-human CML antibody (1:500 dilution)15 or mouse anti-human NOX2 antibody (1:10 dilution)20 for 1 hour. The slides were rinsed with phosphate-buffered saline (PBS) and incubated with a biotin-conjugated rabbit-anti-mouse antibody (1:500 dilution, Dako) for 30 min. After PBS washing, the slides were either incubated with streptavidin–horse radish peroxidase (HRP) complex (1:500 dilution, Dako) for CML staining, or with the ABC kit (1:100 dilution; Vector Lab, Burlingame, California, USA) for NOX2 staining for 1 hour. Following visualisation with 3,3′-diaminobenzidine (Dako) for 8–10 min, the slides were counterstained with haematoxylin, dehydrated in 100% ethanol and covered.
Tissue analysis
The CML staining was quantified using an intensity scoring method, whereby each CML-positive blood vessel was given an intensity score of weak (1), moderate (2) or strong positive (3).27 To obtain the CML IH score, each intensity score was multiplied by the number of blood vessels positive for this score. These were then added and subsequently divided by the surface area of the analysed tissue, resulting in an IH score per square centimetre. The surface area of the tissue was determined using QPRODIT V.3.2 (Leica Microsystems, Cambridge, UK).28 To quantify the NOX2 staining, the number of NOX2-positive blood vessels was counted and divided by the tissue surface area.
Statistical analysis
Data analysis was performed with Prism V.4.0 (GraphPad Software, La Jolla, California, USA). Normal distribution was assessed with a Shapiro-Wilk normality test. Consequently, a parametric unpaired t-test or a one-way ANOVA combined with a Tukey multiple comparison test was employed to analyse differences between groups. Pearson correlation coefficients were calculated for the correlation analyses. Data values are displayed as mean±SE and p<0.05 was considered to be statistically significant for all analyses.
Results
Increased presence of CML and NOX2 in the brain microvasculature after acute MI
CML and NOX2 were found on the endothelium of cerebral microvessels of both control patients and patients with MI (figure 1).

Immunohistochemical staining of CML and NOX2 in the cerebral vasculature. Examples of CML (A) and NOX2 (B) staining in cerebral blood vessels in patients with MI (×200 magnification). Arrows indicate staining of endothelial cells. CML, Nε-(carboxymethyl)lysine; MI, myocardial Infarction; NOX2, NADPH oxidase 2.
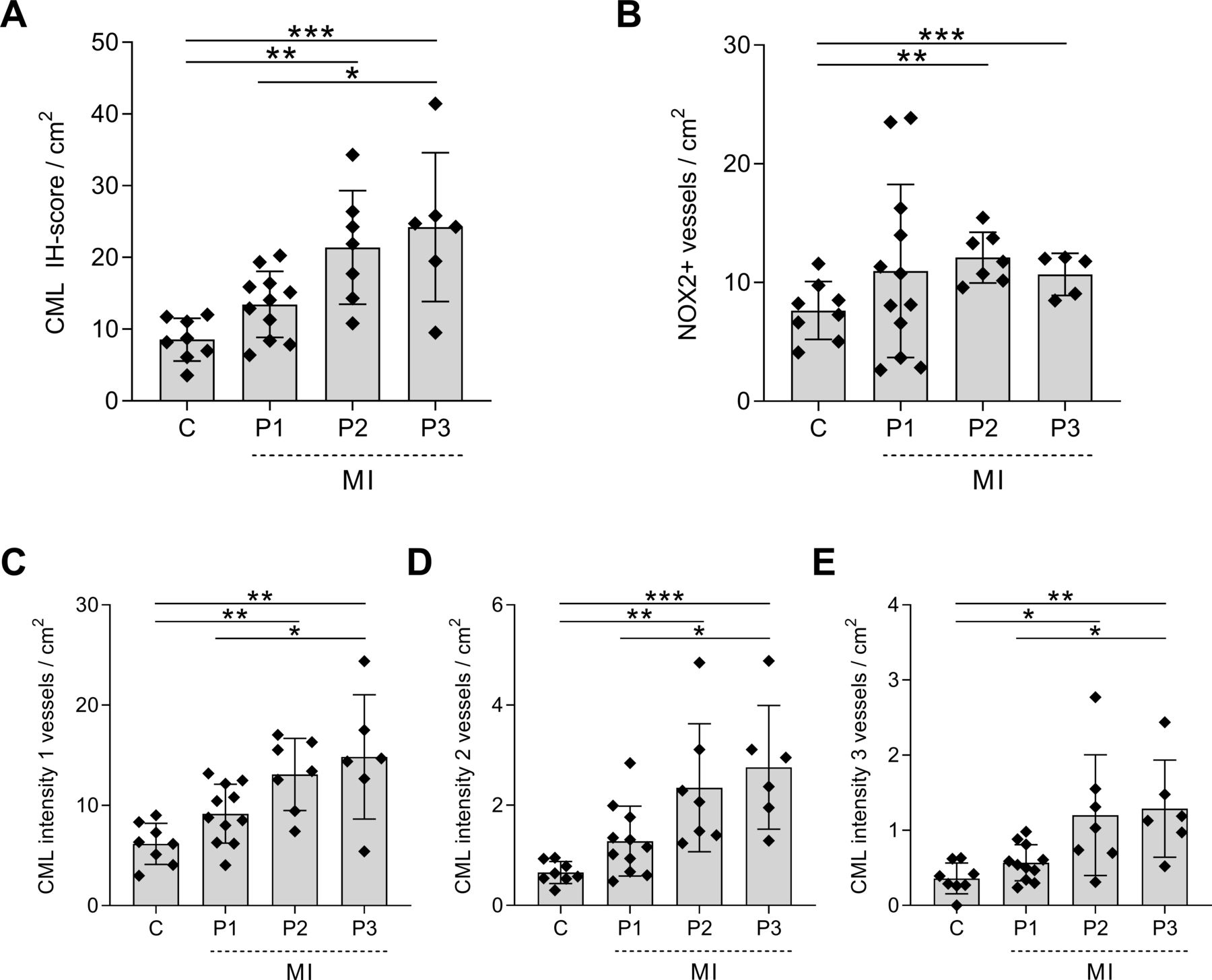
In control patients, the IH score per square centimetre for CML was 8.55±2.98 (figure 2A), which increased to 13.45±4.60 in patients with phase I MI. In both patients with phase II (21.39±7.91, p=0.004) and patients with phase III (24.21±10.37, p=0.0007) MI, the CML IH score per square centimetre was significantly higher compared with controls.
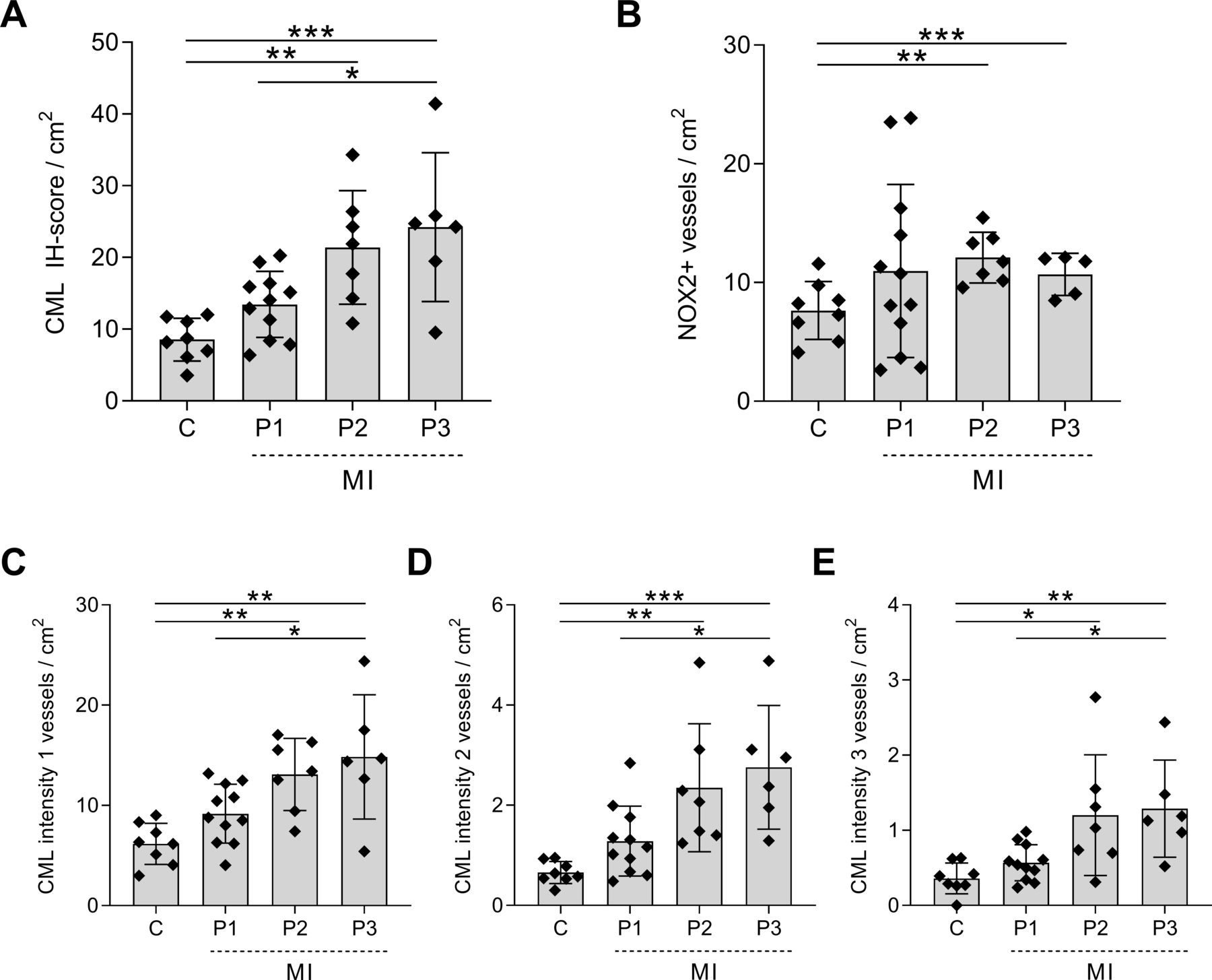
Increased CML and NOX2 expression in the cerebral vasculature of patients with MI. The IH score per square centimetre for CML (A) and NOX2 (B) in the cerebral vasculature of non-MI control patients (C) and patients with MI at different time points after MI: 3–6 hours after MI (P1), 6 hours–5 days after MI (P2) and 5–14 days after MI (P3). The total CML IH score includes all intensities. Separately, weak, score 1 (C); moderate, score 2 (D); and strong, score 3 (E) are depicted. A one-way analysis of variance was used for analysis; data are presented as mean±SE. *P<0.05, **P<0.01, ***P<0.001. CML, Nε-(carboxymethyl)lysine; IH, immunohistochemical; MI, myocardial Infarction; NOX2, NADPH oxidase 2.
We then compared the number of CML-positive blood vessels score per square centimetre with similar staining intensity between the groups. The number of blood vessels with an intensity score of 1 was significantly higher in the last two phases of the MI (phase I: 9.18±2.93, p=0.32; phase II: 13.10±3.60, p=0.006; phase III: 14.84±6.20, p=0.0009) than in control patients (6.16±2.056) (figure 2C). Similarly, the numbers of positive blood vessels with an intensity score of 2 were significantly higher in both phase II (2.35±0.28, p=0.006) and phase III (2.76±1.24, p=0.0009) MI compared with controls (0.66±0.22, figure 2D). Patients with phase II and phase III MI also showed a significantly higher number of blood vessels with an intensity score of 3 (phase II: 1.20±0.80, p=0.01; phase III: 1.29±0.65, p=0.009) than control patients (0.36±0.21, figure 2E).
Patients with MI showed an increase in the number of NOX2-positive blood vessels per square centimetre compared with control patients (7.65±2.44), although this increase was only significant in patients with phase II MI (12.11±2.14, p=0.002) and phase III MI (10.69±1.77, p=0.04) (figure 2B).
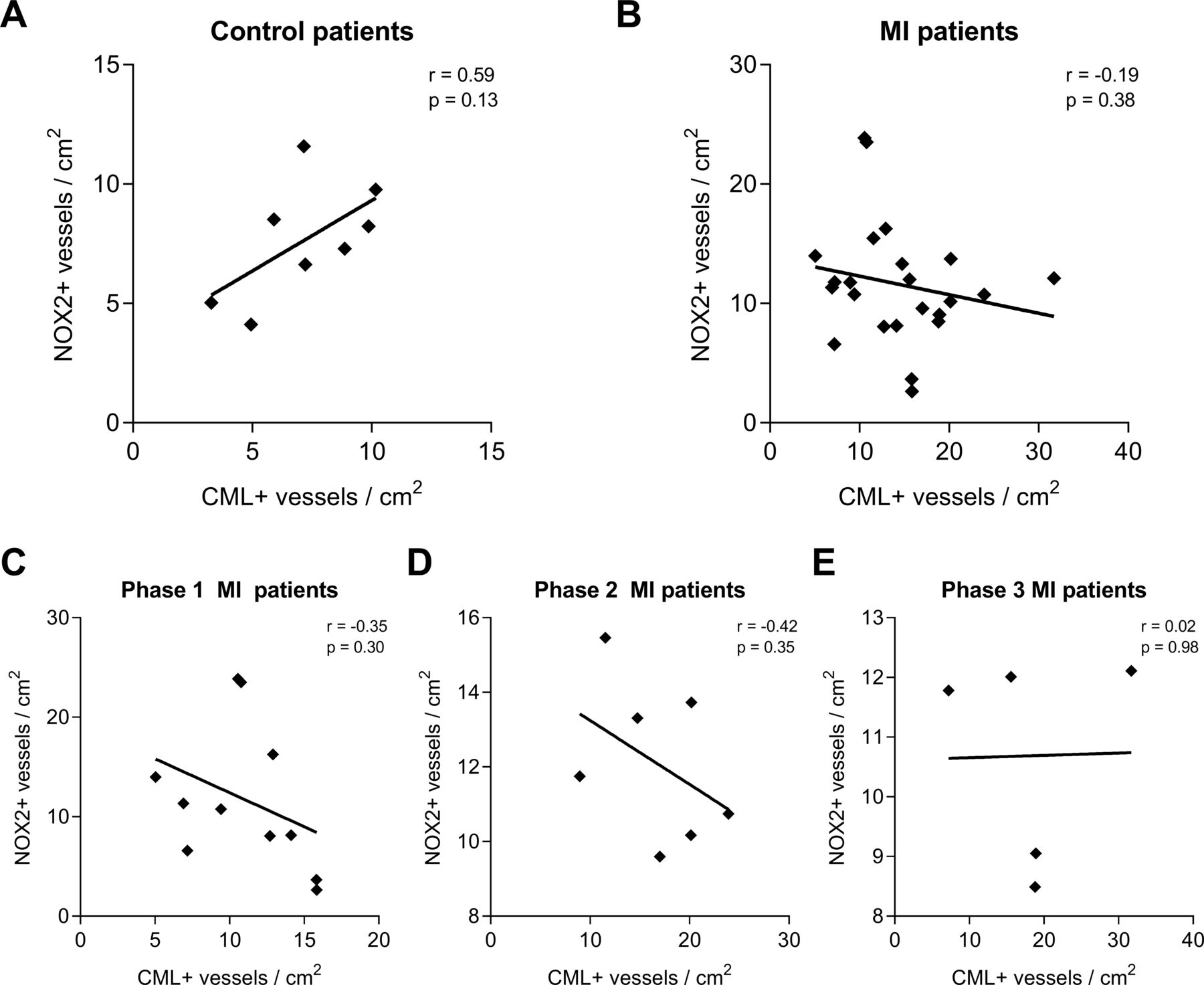
Subsequent correlation analysis of CML-positive and NOX-positive vessels per square centimetre within the control patients showed a moderate positive correlation (r=0.58, p=0.13; figure 3A). When all patients with MI were combined, a weak negative correlation was found (r=−0.19, p=0.38; figure 3B) that persisted in patients with phase I (r=−0.35, p=0.30; figure 3C) and phase II (r=−0.42, p=0.35; figure 3D) MI, but not in patients with phase III (r=0.02, p=0.98; figure 3E) MI.

{kind=link}
{kind=link}
{kind=link}
CML and NOX2 accumulation in the cerebral vasculature show no correlation. The amounts of CML-positive and NOX2-positive blood vessels per square centimetre were tested for correlation within the non-MI control group (A) and the group of patients with MI (B), the latter being further divided into the different phases after MI infarction: 3–6 hours after MI (P1, C), 6 hours–5 days after MI (P2, D) and 5 to 14 days after MI (P3, E). A linear regression was used for analysis. CML, Nε-(carboxymethyl)lysine; IH, immunohistochemical; MI, myocardial Infarction; NOX2, NADPH oxidase 2.
We did not find a correlation between CML and NOX2, suggesting that the formation of AGEs is independent, at least partly, from NOX2.
Discussion
In this study, we found that CML and NOX2 were both increased in the cerebral microvasculature of patients with MI compared with control patients. Furthermore, the data show an increase in CML and NOX2 in time after MI, with the highest levels in phase II, that is, between 6 hours and 5 days after MI. Correspondingly, early after MI, mainly the number of weak CML-positive blood vessels increased, whereas the number of moderate and strong CML-positive blood vessels increased only in later stages of MI. However, no significant correlation was found between the scores for CML and NOX2.
Previously, we found an increased accumulation of CML in the cardiac microvasculature of patients with phase I MI.15 This suggested that in the heart, the increase in microvascular CML may have occurred prior to the MI. Our present findings that the increased presence of microvascular CML in the brain becomes manifest most predominantly in phase II suggest that this increase is likely the result of MI rather than pre-existent in these patients. Moreover, NOX2 accumulation in the cerebral microvasculatutre increased in the different phases of MI.
As to the specific mechanisms that may underlie this increase, we can still only speculate. It has been shown that both CML formation and NOX2 expression are stimulated by conditions of inflammation.17–19 21 As acute MI induces a substantial systemic inflammatory response that results in increased levels of circulating cytokines, acute phase reactants and leucocytes,29 it is plausible that this increased systemic inflammation plays a role in the induced CML formation and NOX2 expression in the brain. Furthermore, MI induces a reduction in cerebral blood flow9 that is also observed in other cardiac conditions (eg, heart failure and cardiac arrest), which may also stimulate cerebral inflammation and oxidative stress.30–33 Moreover, it was shown that blood levels of CML increase after MI.34 Whether increased CML blood levels contributed to the increase of CML and NOX2 in the brain microvasculature remains to be established. Previous studies have shown that CML can activate NOX2-induced ROS production in endothelial cells17 18 and that phagocyte NOX2 knockout prevented the formation of CML on proteins in vitro.19 Indeed, a positive correlation was found in non-MI control patients, although non-significant. In contrast, the weak negative correlation observed in the patients with MI does not correspond with the literature, and we do not have an explanation yet for this anomaly.
Nonetheless, this increased presence of CML and NOX2 could point to dysfunction of the microvasculature of the brain after MI. The consequences of this and whether it is related to the MI-associated depression and dementia remains to be elucidated. However, there is evidence that supports such a relation. It was shown previously that an increase in CML levels and NOX2 expression and activity in the vasculature of the brain occur in different conditions associated with a risk of reduced brain function, such as DM and ageing.22 35 36 Moreover, cerebral NOX2 activity was previously indicated as a crucial driver of CSVD37 and a recent study indeed showed a possible key role for endothelial NOX2 activation in ageing-related cerebral capillary rarefaction and reduced brain function.38 In addition, the extent of CML levels in cerebral vessels was shown to relate to the severity of cognitive impairment in patients with cerebrovascular disease and only minimal Alzheimer’s pathology.24 25
In conclusion, in this study, we show that the AGE-product CML and the ROS producer NOX2 are increased in the microvasculature of the brain after MI, which could point to MI-induced cerebrovascular dysfunction.
Study limitations
The study consists of relatively few patients, specifically in the subgroups, which may underlie the lack of significance in the correlation analysis. However, we do believe that the current numbers provide a good reflection of the population as both men and women of ranging age are included. Furthermore, there was little additional patient information due to the material being autopsy-derived. Hence, other factors that may influence the outcome of the study cannot be excluded.
Data availability statement
All data relevant to the study are included in the article or uploaded as supplementary information.
Ethics statements
Patient consent for publication
Ethics approval
This study was approved by and performed according to the guidelines of the ethics committee of the AUMC, Location VUmc, and conforms to the principles of the Declaration of Helsinki. Use of the leftover material after the pathological examination has been completed is part of the patient contract in our hospital.
References
Footnotes
AK and UB are joint first authors.
Contributors PAJK and HWMN conceived the original idea and supervised the project. SS and CS helped supervise the project. UB and AK carried out the experiments and wrote the manuscript. All authors were involved with critical assessment and interpretation of the data and the final manuscript. PAJK is the guarantor of this manuscript.
Funding This study was financed by Novo Nordisk BV and European Foundation for the Study of Diabetes (grant number PT67543). All other authors reported that they have no relationships to disclose that are relevant to the contents of this paper.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.