Article Text

Abstract
Objective One of the challenges in hypertrophic cardiomyopathy (HCM) is to determine the pathogenicity of genetic variants and to establish genotype/phenotype correlations. This study aimed to: (1) demonstrate that MYBPC3 c.2149–1G>A is a founder pathogenic variant, (2) describe the phenotype and clinical characteristics of mutation carriers and (3) compare these patients with those with the most frequent pathogenic HCM variants: MYBPC3 p.Arg502Trp/Gln.
Methods We reviewed genetic tests performed in HCM probands at our institution. We carried out transcript analyses to demonstrate the splicing effect, and haplotype analyses to support the founder effect of MYBPC3 c.2149–1G>A. Carriers with this mutation were compared with those from MYBPC3 p.Arg502Trp/Gln in terms of presentation features, imaging and outcomes.
Results MYBPC3 c.2149–1G>A was identified in 8 of 570 probands and 25 relatives. Penetrance was age and sex dependent, 50.0% of the carriers over age 36 years and 75.0% of the carriers over 40 years showing HCM. Penetrance was significantly higher in males: in carriers older than 30 years old, 100.0% of males vs 50.0% of females had a HCM phenotype (p=0.01). Males were also younger at diagnosis (32±13 vs 53±10 years old, p<0.001). MYBPC3 c.2149–1G>A resulted in an abnormal transcript that led to haploinsufficiency and was segregated in two haplotypes. However, both came from one founder haplotype. Affected carriers showed a better functional class and higher left ventricular ejection fraction (LVEF) than patients with MYBPC3 p.Arg502Trp/Gln (p<0.05 for both). Nevertheless, the rate of major adverse outcomes was similar between the two groups.
Conclusions MYBPC3 c.2149–1G>A splicing variant is a founder mutation. Affected males show an early onset of HCM and with higher penetrance than women. Carriers show better functional class and higher LVEF than MYBPC3 p.Arg502Trp/Gln carriers, but a similar rate of major adverse outcomes.
- cardiomyopathy
- hypertrophic
- genetics
- diagnostic imaging
Data availability statement
Data are available on reasonable request. All data relevant to the study are included in the article or uploaded as online supplemental information.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Key questions
What is already known about this subject?
Hypertrophic cardiomyopathy (HCM) is the most common inherited cardiac disease, and it is genetically and clinically heterogeneous. Thousands of pathogenic variants causing HCM have been described but few genotype–phenotype correlations have been established because most of the pathogenic variants are unique or seen in a limited number of families.
What does this study add?
This study shows that MYBPC3 c.2149–1G>A is a founder mutation that alters splicing, changes the reading frame and causes a truncated protein. The cosegregation of MYBPC3 c.2149–1G>A in eight non-related HCM families confirms the pathogenicity of the variant. Most MYBPC3 c.2149–1G>A carriers present a moderate left ventricular hypertrophy, but a highly variable expressivity is observed. The incidence of major adverse outcomes is similar to other HCM variants. Male carriers show an earlier onset and a higher penetrance than female carriers.
How might this impact on clinical practice?
The confirmation of MYBPC3 c.2149–1G>A pathogenicity permits restricting the follow-up of relatives to mutation carriers. These will need lifetime surveillance to early identify the development of disease and treat and prevent its complications.
Introduction
Hypertrophic cardiomyopathy (HCM) is the most common inherited cardiac disease with an estimated prevalence of 1 in 500 individuals.1 HCM is a heterogeneous disease with a wide clinical spectrum, characterised by increased left ventricular wall thickness (LVWT) in the absence of abnormal loading conditions.1 2 Although many patients have asymptomatic or mildly symptomatic forms and normal life expectancy, other patients develop malignant phenotypes associated with sudden cardiac death (SCD) and end stage heart failure. Atrial fibrillation (AF) and left ventricular outflow tract obstruction (LVOTO) are additional common complications.3 HCM is inherited with an autosomal dominant pattern, with incomplete and age dependent penetrance. A pathogenic variant in a sarcomere gene is found in about 35%–50% of HCM cases and MYBPC3 is the most frequent affected gene.4–6
More than 2000 pathogenic variants in sarcomeric genes have been described until date. Most of them have a very low frequency in HCM cohorts or are confined to individual families.3 7 An exception is the MYBPC3 p.Arg502Trp variant (c.1504G>T). Representing 1.5%–3% of HCM patients, this mutation is the most frequent in different HCM cohorts.4 7 8 Two other missense pathogenic variants have been described at this position, p.Arg502Gln (c.1505G>A) and p.Arg502Leu (c.1505G>T).9
Few specific genotype–phenotype correlations have been demonstrated in HCM, probably because most of the pathogenic variants are unique or seen in limited numbers. However, when founder pathogenic mutations are identified, their study represents a unique opportunity to describe clinical phenotypes.10–19 The MYBPC3 c.2149–1G>A variant has been previously described in a single HCM proband, without clinical and segregation data.20 In the ClinVar public database, this variant is reported in other three non-related HCM patients, but there is still no clinical or segregation information available. The sequence change affects an acceptor splice site, so it is expected to result in an absent or disrupted protein. However, no functional analyses have been conducted. The main objectives of this study were (1) to address whether the identified MYBPC3 c.2149–1G>A mutation is a founder pathogenic variant, (2) clarify its functional consequences and (3) to describe clinical characteristics and phenotype of affected carriers. In addition, we compared the phenotype and clinical profile of patients with this mutation with a control group of MYBPC3 p.Arg502Trp/p.Arg502Gln carriers, the most frequent HCM pathogenic variants in our institution.
Methods
Study population and genetic analysis
We reviewed all genetic analyses in HCM probands performed in our institution between January 2012 and August 2019, screening for MYBPC3 variants. Genetic analyses in HCM probands were performed by Sanger sequencing of the five main sarcomeric genes (MYBPC3, MYH7, TNNT2, TNNI3 y TPM1) between January and November 2012, and by next-generation sequencing of a panel of 25 HCM-related genes from that date onwards. All patients signed an informed consent document authorising the use of their genetic data for research purposes.
Patient and public involvement
Patients were not involved in the design, development of the study or the interpretation and writing of the results.
Clinical evaluation
Following current recommendations,1 2 the diagnosis of HCM was established by a left ventricular hypertrophy ≥15 mm in probands and ≥13 mm in relatives. A pedigree was elicited for each proband, and relatives were offered clinical and genetic screening if a pathogenic or likely pathogenic mutation was identified in the proband. HCM patients were followed up and treated according to current recommendations. We defined LVOTO as the presence of a left ventricle outflow tract pressure gradient ≥30 mm Hg at rest. A pressure gradient ≥50 mm Hg was considered haemodynamically relevant.1 2 All HCM patients, whether they were gene positive or negative, underwent arrhythmic risk stratification based on the presence of recognised arrhythmic risk factors for HCM. Since 2014, the 5-year risk of SCD was estimated according to the European Society of Cardiology (ESC) score.1 21 Any episode of ischaemic stroke, SCD, aborted sudden cardiac arrest (SCA), appropriate implantable cardioverter defibrillator (ICD) shock, heart failure admission, cardiac transplantation or cardiac death was recorded as a major adverse outcome.
Cardiovascular magnetic resonance
All probands and relatives that fulfilled the HCM criteria at first evaluation were offered scanning by cardiovascular MR (CMR). Patients underwent CMR with a 1.5-T Philips Achieva scanner. The CMR study consisted of cine steady-state free-precession imaging of left ventricular function and late enhancement imaging of myocardial scar tissue (3D inversion-recovery turbo gradient echo sequence). Images were obtained in short-axis views and four-chamber, two-chamber and three-chamber views. Late gadolinium enhancement (LGE) was performed 10 min after a total injection of 0.2 mmol/kg gadoteridol. CMR data were analysed by investigators blinded to patient’s genotype, using dedicated software (QMass MR V.8.1, MEDIS Suite V.3.2). Left ventricular end-diastolic and end-systolic volumes, ejection fraction and mass were measured from short axis views.22 The presence of LGE was determined by visual inspection. Scar size (extent) was calculated from LGE sequences as a percentage of myocardium by with semi-automated planimetry (manually corrected) using full-width half-max thresholding.
Splicing analysis
In silico analysis of MYBPC3 c.2149–1G>A was performed using Human Splicing Finder (http://www.umd.be/HSF/HSF.shtml) and MaxEntScan (http://hollywood.mit.edu/burgelab/maxent/Xmaxentscan_scoreseq.html) bioinformatic prediction tools. Additionally, RNA isolated from blood samples of one MYBPC3 c.2149–1G>A carrier and one non MYBPC3 c.2149–1G>A carrier was analysed by PCR using a primer pair from exon 22 to exon 24 (Fw: 5’-AGCCCCAGATGCCCCAGAGGA-3’and Rv: 5’-GTAGGCAGGCGGCTCCCACTGTA-3’), to validate transcript sizes.
Ancestor analysis
To identify the haplotypes sharing MYBPC3 c.2149–1G>A variant, six markers: rs7120013, rs4882135, rs671299, rs7116652, rs10792299 and rs11605489 (online supplemental table S1) and MYBPC3 c.2149–1G>A variant were genotyped by Sanger sequencing in available samples, 16 MYBPC3 c.2149–1G>A carriers and 6 non-carriers. These markers covered from 46 to 62 Mb of chromosome 11, and segregate at intermediate frequency at population level (selected from the 1000 Genomes Browser database, www.ncbi.nlm.nih.gov/variation/tools/1000genomes/). Genotype information for those markers was extracted for an Iberian population (n=107) from the 1000 Genomes Browser database. Haplotypes were reconstructed employing family information and the Iberian population genotype data to infer most probable haplotypes using phase V.2.1 software.23 Reconstructed haplotypes were subjected to phylogenetic analysis using minimum evolution, bootstrap (1000 replicates) and tree neighbor-joining in MEGA V.7 software.24
Supplemental material
Statistical analysis
Data are expressed as mean±SD and frequencies or percentages where appropriate. We used ordinary least-square linear regression to compare continuous variables, and χ2 to compare qualitative variables. The penetrance, diagnosis age and cumulative probability of an event on follow-up was estimated using the Kaplan-Meier method. We used log-rank tests and Cox proportional-hazards models to compare survival between groups (MYBPC3 variant and gender). A p<0.05 was considered statistically significant.
Results
Study population
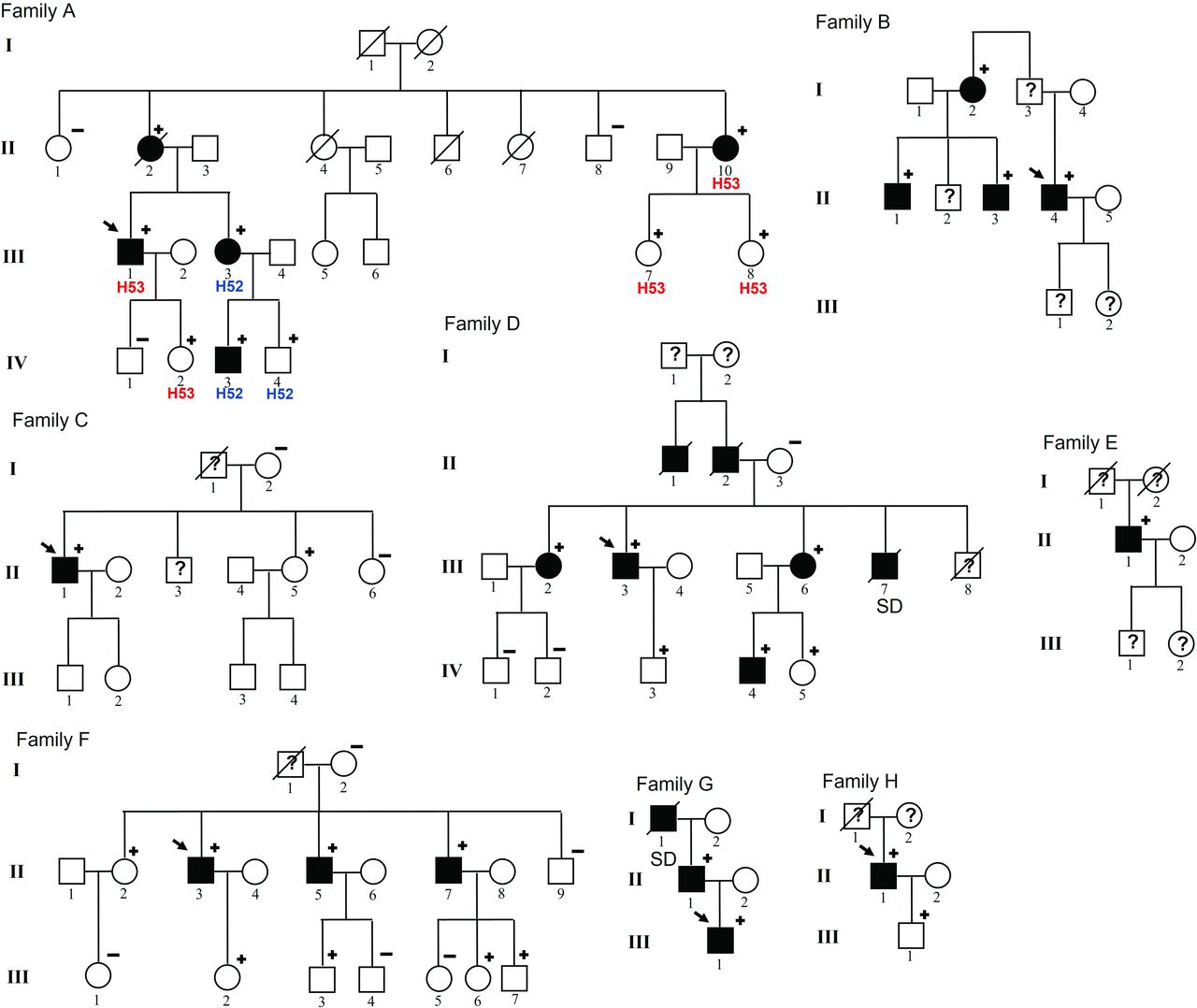
A total of 570 HCM probands underwent genetic testing at our institution between 2012 and 2019. A MYBPC3 pathogenic variant was identified in 79 (14.0%). MYBPC3 p.Arg502Gln (c.1505G>A, NM_00256.3) and MYBPC3 p.Arg502Trp (c.1504C>T, NM_00256.3) were the most frequent variants detected in MYBPC3, identified in heterozygosis in 16 unrelated HCM probands (2.8% of the HCM probands) and 17 relatives (8 were affected, 47.0%). The MYBPC3 c.2149–1G>A (NM_00256.3) variant was identified in eight unrelated HCM probands (all male), representing 1.4% of all probands. We studied 45 family members and identified 25 MYBPC3 c.2149–1G>A carriers, 12 (48.0%) of them with HCM phenotype and 13 (52.0%) unaffected carriers (figure 1).

MYBPC3 c.2149-1G>A pedigrees. Symbols denote sex, genetic and disease status: +, carriers; –, non-carriers; ?, unknown phenotype; box, male; circle, female; darkened, affected; slashed, deceased; clear symbol, unaffected; without sign, not studied. SD, sudden death.
Penetrance and age at diagnosis
Overall penetrance in MYBPC3 c.2149–1G>A carriers was 62.0% and it was age dependent: 50.0% of the carriers over age 36 years and 75.0% of the carriers over 40 years had HCM phenotype. Moreover, in male patients older than 30 years, penetrance was 13/13 (100.0 %), significantly higher than in females above this age (5/10, 50.0%, p=0.01). The mean age at diagnosis was 37±15 years, significantly lower in males than in females (32±13 vs 53±10, respectively, p<0.001). There were no significant differences on penetrance and age at diagnosis compared with MYBPC3 p.Arg502Trp/Gln carriers (log-rank p=0.16 and p=0.60 for penetrance and age at diagnosis, respectively). For MYBPC3 p.Arg502Trp/Gln, age at diagnosis was also lower in males (28±20 vs 50±19 years old, p=0.007). However, as opposed to MYBPC3 c.2149–1G>A carriers, no gender differences (p=0.08) on penetrance were observed for carriers of this control variant (table 1 and figure 2).

Penetrance. Comparison between MYBP3 c.2149–1G>A and MYBPC3 p.Arg502Trp/Gln variants and between sex for each variant. (A) Full study population. (B) MYBP3 c.2149–1G>A sex analysis. (C) MYBPC3 p.Arg502Trp/Gln sex analysis. HCM, hypertrophic cardiomyopathy.
MYBPC3 c.2149–1G>A and MYBPC3 p.Arg502Tr/Gln comparison
Phenotype, clinical characteristics and outcomes
Phenotype and clinical data from 20 identified MYBPC3 c.2149–1G>A carriers were used for comparison. The reason for diagnosis were symptoms in 6 patients (30.0%), and familiar screening or casual finding in 14 (70.0%). At first evaluation, 19 affected carriers (95.0%) showed asymmetric left ventricular hypertrophy affecting the anterior septum, only one patient presented a concentric LVH pattern. Mean LVWT was 21±6 mm and 19±5 mm measured by echocardiography and CMR, respectively. The left atrium was enlarged in 12 of 20 (60.0%) affected carriers. LVOTO at rest was detected in three individuals at diagnosis, being haemodynamically relevant in two cases, although to date none of them has required invasive management (table 1). CMR was performed in 18 of the 20 affected carriers, showing LGE in 15 of them (83%). The three patients without LGE were young patients with mild phenotypes (maximum LVWT ≤16 mm). Mean LV mass was 65±29 g and mean percentage of fibrosis was 16%±14, with 35.0% of patients showing a percentage of fibrosis >15% (table 2).
MYBPC3 c.2149–1G>A and MYBPC3 p.Arg502Tr/Gln cardiac MR data
A great variability on phenotype was detected in patients carrying MYBPC3 c.2149–1G>A that varied from normal phenotype to severe disease with extensive fibrosis or even restrictive phenotype (figure 3A–H).

Cardiac MR (CMR) images showing phenotype variability in MYBPC3 c.2149-1G>A carriers. (A–D) CMR images of two brothers: A, B is a 48-year-old female carrier with normal phenotype and C, D is her 49-year-old affected brother with septal hypertrophy of 22 mm and extensive LGE. (E, F) CMR images showing a severe left ventricle hypertrophy with extensive LGE in a 21-year-old carrier with severe systolic disfunction. (G, H) CMR images showing a restrictive phenotype with severe atrial enlargement and fibrosis. LGE, late gadolinium enhancement.
Follow-up data for MYBPC3 c.2149–1G>A affected patients was available for a median of 9 (range 1–34) years since diagnosis. After this period, 19 patients (95.0%) remained mildly symptomatic (New York Heart Association I–II). Four patients (20.0%, 4 males) developed AF. At least an episode of non-sustained ventricular tachycardia was detected in 7 patients (35.0%). Nine patients (45.0%) had a major classical SCD risk factor (six had a family history of SCD, two had LVWT≥30 mm, one had a syncope). The ESC risk score was higher than 4% in eight carriers (40.0%). These eight patients were offered an ICD which was finally implanted in seven (35.0%) because of patient’s preferences. One of the ICD implants took place in secondary prevention after an aborted SCA, as the patient had also previously rejected ICD implantation (table 1).
There were six major adverse outcomes registered: one patient suffered an aborted SCA and posteriorly appropriate ICD shocks, and another patient had appropriate ICD shocks. Two patients were admitted for heart failure and another patient with a restrictive phenotype developed advanced heart failure and underwent evaluation for cardiac transplantation (table 3). The rate of major adverse outcomes was 0.82% per year.
Major clinical outcomes in MYBP3 c.2149–1G>A and MYBPC3 p.Arg502Trp/Gln affected carriers
Compared with MYBPC3 p.Arg502Trp/Gln, MYBPC3 c.2149–1G>A patients had higher left ventricular ejection fraction (LVEF) (p=0.03) and better functional class (p=0.04; table 1). No significant differences for CMR measures were identified between both MYBPC3 variants (table 2). Survival analysis showed similar outcomes in both patient groups (HR 0.3, (95% CI 0.3 to 4.9) p=0.706; table 3 and figure 4).
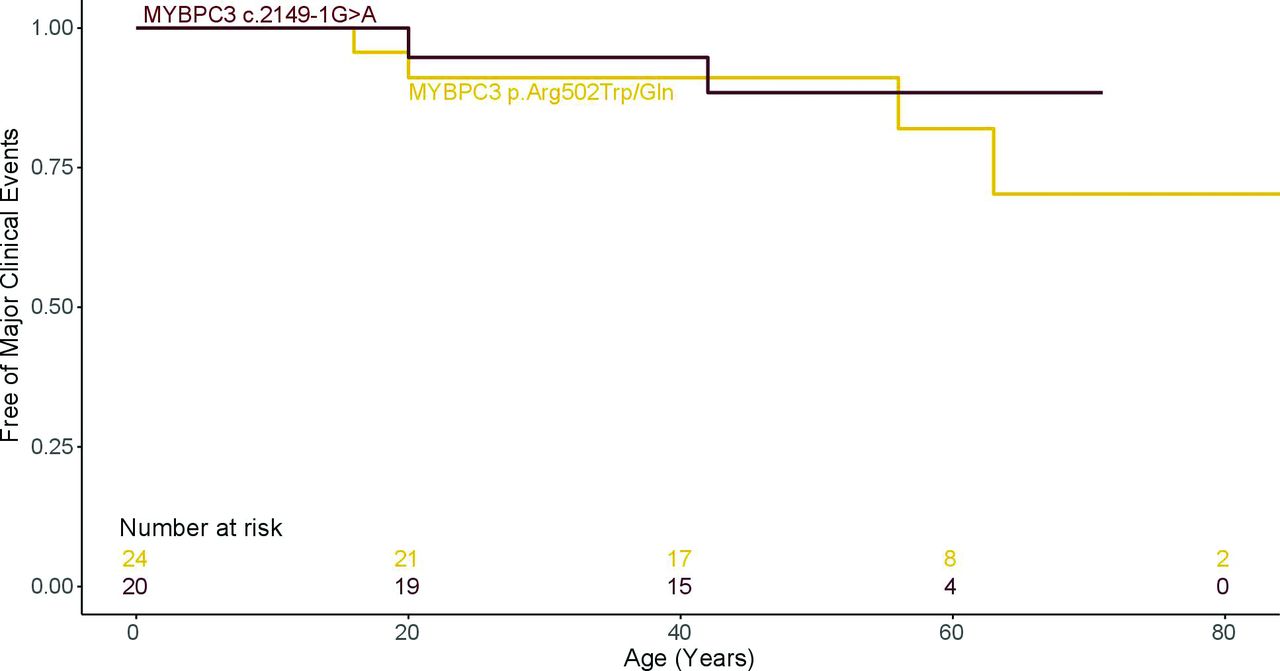
Survival free from major clinical outcomes. Comparison between MYBPC3 c.2149–1G>A and MYBPC3 p.Arg502Trp/Gln variants.
Splicing analysis
HSF and MaxEntScan tools predicted that MYBPC3 c.2149–1G>A alters the acceptor splice site producing an abnormal splicing. In this case, intron between exon 22 and 23 would skip splicing and would be included in the mature RNA, changing the reading frame and causing a truncated protein. Transcript analysis confirmed that MYBPC3 c.2149–1G>A generated an abnormal transcript (online supplemental figure S1).
Supplemental material
Ancestor analysis
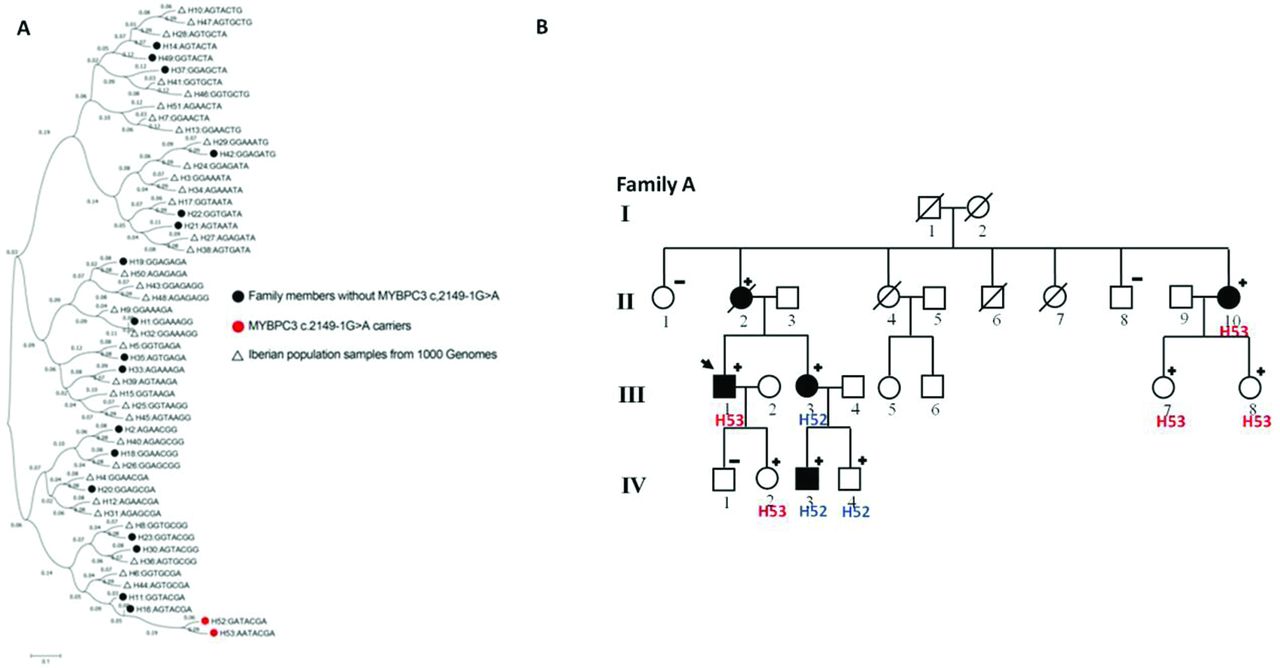
The haplotype reconstruction showed 53 different haplotypes in the Iberian population. The most probable segregation for MYBPC3 c.2149–1G>A carriers was estimated in two haplotypes, H52: GATACGA and H53: AATACGA. The phylogeny reconstruction estimated that both H52 and H53 may come from common ancestor H16 (figure 5A). In fact, H52 may come from one recombination event in H53 haplotype that has recently occurred in one of the families (figure 5B).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Haplotype reconstruction analysis. (A) phylogeny reconstruction of MYBPC3 haplotypes. Tree Neighbor-Joining in MEGA7. (B) segregation of H52 and H53 haplotypes in a family carrying MYBPC3 c.2149–1G>A.
Discussion
To our knowledge, this is the first study to describe the clinical phenotype of the MYBPC3 c.2149–1G>A variant, herein demonstrating cosegregation in eight non-related HCM families (figure 1). This information confirms the pathogenicity of the variant. Hence, this variant can be classified as a definite pathogenic variant with strong evidence according to the American College of Medical Genetics criteria.25 This information is essential for patient management because it enables to perform genetic screening in family members and narrows the clinical follow-up to be performed only in carriers.
Mechanism of pathogenicity and founder effect
MYBPC3 c.2149–1G>A alters the canonical donor splice sequence, so it is supposed to generate alternative splicing. However, to our knowledge, no functional studies on this mutation are available. This is the first time that RNA analysis is performed, demonstrating that this variant alters normal splicing. This G>A change triggers intron 22–23 retention, causing a frameshift that leads to a truncated protein. Unlike other sarcomeric genes, where the majority of pathogenic variants are missense with a dominant negative effect, up to 60% of the pathogenic variants in MYBPC3 are frameshifts leading to haploinsufficiency.26 In fact, all the founder pathogenic variants previously described in MYBPC3 are truncated variants.10–19 On the other hand, the mechanisms of pathogenicity of MYBPC3 p.Arg502Trp and p.Arg502Gln are unclear. Previous studies have shown that MYBPC3 p.Arg502Trp alters the predicted electrostatic properties of the C3 domain of the protein and may directly disrupt the interaction of MYBPC3 with other sarcomeric proteins.27
The haplotype analyses performed in our study supports a founder origin of MYBPC3 c.2149–1G>A, as all the analysed carriers, related and unrelated, shared the same haplotype along 16 Mb. Actually, two haplotypes carrying MYBPC3 c.2149–1G>A were identified in one single family, which likely corresponds to a recent recombination event as all other family members share the same MYBPC3 c.2149–1G>A haplotype. All carriers are from the same geographical area, the South region of Madrid (Spain). The opposite to founder mutation occurs with MYBPC3 p.Arg502Trp variant, where several different haplotypes appear segregating with the variant.9 Therefore, the high frequency of this variant seems to be explained by recurrent mutation of this residue rather than a founder effect. Further evidence that supports that MYBPC3 codon 502 may be a mutational hotspot is the existence of two other HCM mutations that alter arginine 502 to either glutamine or to leucine and the fact that variants at this position are frequent in HCM cohorts from different countries.4 7–9
Penetrance, phenotype and clinical characteristics
Although the penetrance of MYBPC3 c.2149–1G>A was incomplete and age-dependent as in most MYBPC3 variants, penetrance was particularly high in young males, reaching 100.0% in carriers older than 30 years, and significantly higher in males than females. Also, males were younger at the time of diagnosis. Similar gender differences have been described for other MYBPC3 variants.17–19 28 This delay in developing the disease in women may be secondary to genetic and endocrine factors that may influence phenotypic expression. Other suggested explanations are the lack of attention to early clinical signs and fewer indications for medical screening programmes in women.28 In our study, all affected female carriers of MYBPC3 c.2149–1G>A were diagnosed after the age of 40 during family screening and most of them were asymptomatic at diagnosis. Therefore, the older age at diagnosis in females in our cohort is better explained by a later-age onset disease than by a delayed diagnosis.
Our imaging data show that MYBPC3 c.2149–1G>A patients present an HCM phenotype with left ventricular thickening affecting anterior septum, preserved LVEF, frequent left atrial enlargement and extensive LGE with 35.0% of patients showing relevant fibrosis. However, expressivity was highly variable, ranging from frequent non-affected carriers all the way to a few patients with a severe phenotype and adverse outcomes. This observation may support the relevance of modifier genes and epigenetics and internal/external environmental factors that still are not well understood.29
MYBPC3 c.2149–1G>A affected carriers showed higher LVEF and a better functional class compared with p.Arg502Trp/Gln affected carriers. However, the rate of major adverse outcomes was similar between the two groups. It is believed that founder mutations give rise to benign or intermediary phenotypes and disease course to withstand negative selection pressure, most of the carriers surviving the reproductive age. This allows these variants to perpetuate through multiple generations.30 Nevertheless, 20.0% of MYBPC3 c.2149–1G>A affected carriers showed a severe phenotype. Also, the proportion of patients in our cohort showing a high estimated risk of SCD (40.0% according to ESC risk score, 50.0% according to American College of Cardiology/American Heart Association guidelines) as well as the proportion of patients undergoing ICD implantation (35.0%) and incidence of appropriate ICD shocks (10.0%) is higher than described in other recent HCM cohorts.15 17 31 Hence, the MYBPC3 c.2149–1G>A variant should not be considered a benign mutation with a mild phenotype. Although variants affecting the position 502 of MYBPC3 are a frequent cause of HCM worldwide, data relating phenotype and clinical outcomes is scarce.8 9 Future studies with larger patient cohorts would help clarify genotype-phenotype relations for these frequent variants.
Limitations
The study is limited by the relatively small numbers of MYBPC3 c.2149–1G>A affected carriers, which challenges to describe a conclusive genotype–phenotype relation for this pathogenic variant.
Conclusions
MYBPC3 c.2149–1G>A is a founder pathogenic variant generating an alternative splicing that leads to a truncated MYBPC3 protein. Male carriers show an early onset and a high penetrance of HCM. Carriers show better LVEF and functional class than MYBPC3 p.Arg502Trp/Gln carriers but have a similar rate of major adverse outcomes.
Data availability statement
Data are available on reasonable request. All data relevant to the study are included in the article or uploaded as online supplemental information.
Ethics statements
Patient consent for publication
Ethics approval
The study was approved by the Ethics Committee of the Hospital General Universitario Gregorio Marañón (Madrid, Spain) and performed in compliance with the Declaration of Helsinki.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors IMF, AIF, MÁE, SC, RL, RY, FFA and JB contributed to the design of the study. IMF, JFR, MAT and MG-M collected the data. AIF performed the statistical analysis, the molecular and haplotype analysis and the bioinformatic predictions. All authors contributed to the interpretation of the data and the review of manuscript drafts, and all approved the final manuscript. The corresponding author attests that all listed authors meet authorship criteria and that no others meeting the criteria have been omitted.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.