Article Text

Abstract
Aims To phenotype patients referred to a tertiary centre for the exploration of a left ventricular hypertrophy (LVH) starting from 12 mm of left ventricular wall thickness (LVWT).
Methods and results Consecutive patients referred for aetiological workup of LVH, beginning at 12 mm of LVWT were retrospectively included in this tertiary single-centred observational study. Patients presenting with severe aortic stenosis were excluded. Aetiological workup was reviewed for each subject and aetiologies were adjudicated by expert consensus.
Among 591 patients referred for LVH aetiological workup, 41% had a maximal LVWT below 15 mm. LVH aetiologies were led by cardiac amyloidosis (CA, 34.3%), followed by sarcomeric hypertrophic cardiomyopathy (S-HCM, 32.1%), hypertensive cardiomyopathy (21.7%), unknown aetiology (7.6%) and other (4.2%), including Anderson-Fabry’s disease (1.7%). CA and S-HCM affected over 50% of patients with mild LVH (12–14 mm); the prevalence of these aetiologies rose with LVH severity. Among patients with Anderson-Fabry’s disease, 4 (40%) had a maximal LVWT <15 mm.
Conclusions Mild LVH (ie, 12–14 mm) conceals multiple aetiologies that can lead to specific treatment, cascade family screening and specific follow-up. Overall, CA is nowadays the leading cause of LVH in tertiary centers.
- cardiomyopathies
- cardiomyopathy
- hypertrophic
- epidemiology
- genetic diseases
- inborn
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Key questions
What is already known about this subject?
Current European guidelines define hypertrophic cardiomyopathy (HCM) as an increased left ventricular wall thickness (LVWT) ≥15 mm that is not explained by loading conditions.
This thickness cutoff is arbitrary and solely based on the methods of historical studies on HCM.
The admitted distribution of HCM aetiologies mainly relies on genetic studies and is about 60% sarcomere gene mutation, 30% unknown and 10% other.
What does this study add?
Many patients exhibit only mild left ventricular hypertrophy (LVH) (ie, LVWT 12–14 mm).
Multiple meaningful LVH aetiologies are concealed in this group.
Overall, amyloidosis is an increasing cause of LVH.
How might this impact on clinical practice?
The definition of HCM could be revised.
The threshold to initiate explorations could be lowered to allow early detection, especially considering the emergence of specific treatments for common LVH aetiologies.
Introduction
Current 2014 guidelines for the diagnosis and management of hypertrophic cardiomyopathies (HCMs) from the European Society of Cardiology1 define HCM in adults as ‘a wall thickness (WT) ≥15 mm in one or more left ventricular (LV) myocardial segments—as measured by any imaging technique (echocardiography, cardiac magnetic resonance imaging (CMR) or CT)—that is not explained solely by loading conditions’. This threshold is set on the basis of historical studies2 in the field of HCMs in which an arbitrary value of 15 mm was used. However, there is no scientific rationale for this cut-off value. Studies carried out in healthy subjects3 have shown that the normal LVWT range is 6–11 mm.
The admitted distribution of HCM aetiologies (relying on various, mostly genetic-based, studies) is presented as such: 40%–60% sarcomeric protein gene mutations, 25%–30% unknown and 5%–10% genetic and non-genetic causes.1 This last category aggregates numerous and diverse aetiologies: inborn errors of metabolism, neuromuscular diseases, mitochondrial diseases, malformation syndromes, amyloidosis, newborn of diabetic mother and drug-induced HCMs.
Clinical practice challenges these statements. First, there seem to exist many HCM diagnoses below the 15 mm cut-off. Second, real-life distribution of HCM aetiologies seems to diverge from the one of the guidelines.
The aim of this study was to phenotype patients referred to a tertiary centre for the exploration of a LV hypertrophy (LVH) starting from 12 mm of LVWT.
Methods
Study population and data collection
The medical records of all consecutive patients referred to our tertiary University Hospital, Department of cardiac investigations, Toulouse, France, for the diagnostic work-up of LVH were retrospectively reviewed from January 2015 to July 2019. To accurately describe this population, a lower LVWT cut-off value than the one defined in the upmentioned guidelines was used and all subjects with a maximal LVWT ≥12 mm as measured by transthoracic echocardiography (TTE) were included in the study. Patients presenting with severe aortic valve stenosis (AS), bioprosthetic aortic valve stenotic degeneration and obstructive subaortic membranes were excluded. Patients with a definite LVH aetiology have been subsequently divided into three tertiles allowing a three-stage LVH gradation: mild (maximal LVWT 12–14 mm), moderate (maximal LVWT 15–16 mm) and severe (maximal LVWT ≥17 mm).
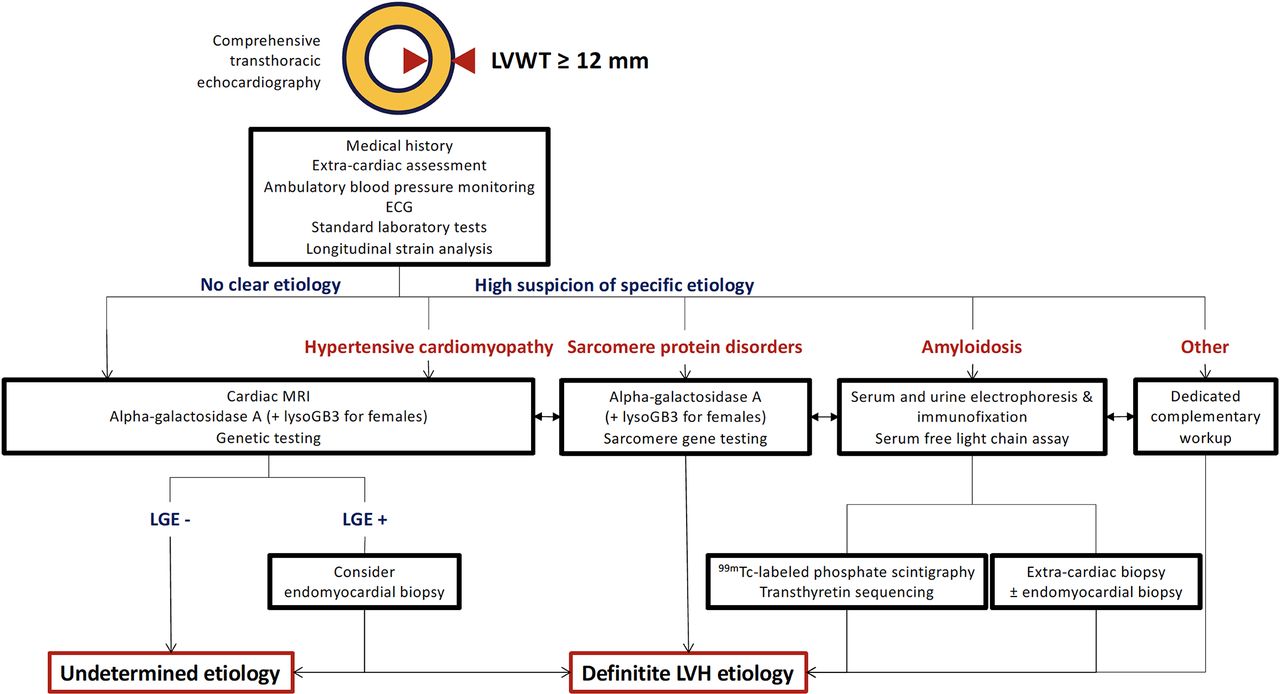
Medical records of all patients were comprehensively reviewed to collect clinical, electrocardiographic, laboratory, imaging and clinical pathology data. The workup algorithm used in our centre is presented in figure 1. Based on these data, we collegially adjudicated the aetiology of each patient’s LVH. For the specific diagnosis of hypertensive cardiomyopathy (HTN-CMP), the following criteria were used: (1) elevated blood pressure at two or more distinct timepoints, (2) at least two antihypertensive medications prescribed and (3) exclusion of other LVH aetiologies.

Workup algorithm for left ventricular hypertrophy aetiology determination. ‘Other’ notably includes inborn errors of metabolism, glycogen storage diseases, neuromuscular diseases, mitochondrial diseases and RASopathies. LGE, late gadolinium enhancement; LVH, left ventricular hypertrophy; LVWT, left ventricular wall thickness; lysoGB3, lyso-globotriaosylsphingosine.
The investigation conforms with the principles outlined in the Declaration of Helsinki. All patients were informed at the admission that their clinical data could be used for research purpose and gave their consent.
TTE review
All TTEs had been performed on General Electric ultrasound systems (General Electric Healthcare, Boston, Massachusetts, USA). All TTE loops were reviewed by a trained cardiologist to assess LV walls and chamber dimensions and left and right ventricle systolic function using EchoPAC Software v202 R34.0 (General Electric, Boston, Massachusetts, USA). Two-dimensional measurements were performed unless LV alignment allowed M-mode measurements.
Statistical analysis
Continuous variables were presented as medians with IQR. Categorical variables were expressed as numbers and percentages. Count data were compared using Fisher’s exact tests or Pearson’s χ2 tests when applicable. A bilateral p<0.05 was considered statistically significant. In post-hoc analyses, p values were adjusted with Holm’s method. All statistical tests were performed using the R software V.3.6.2 (R Foundation for Statistical Computing, Vienna, Austria).
Results
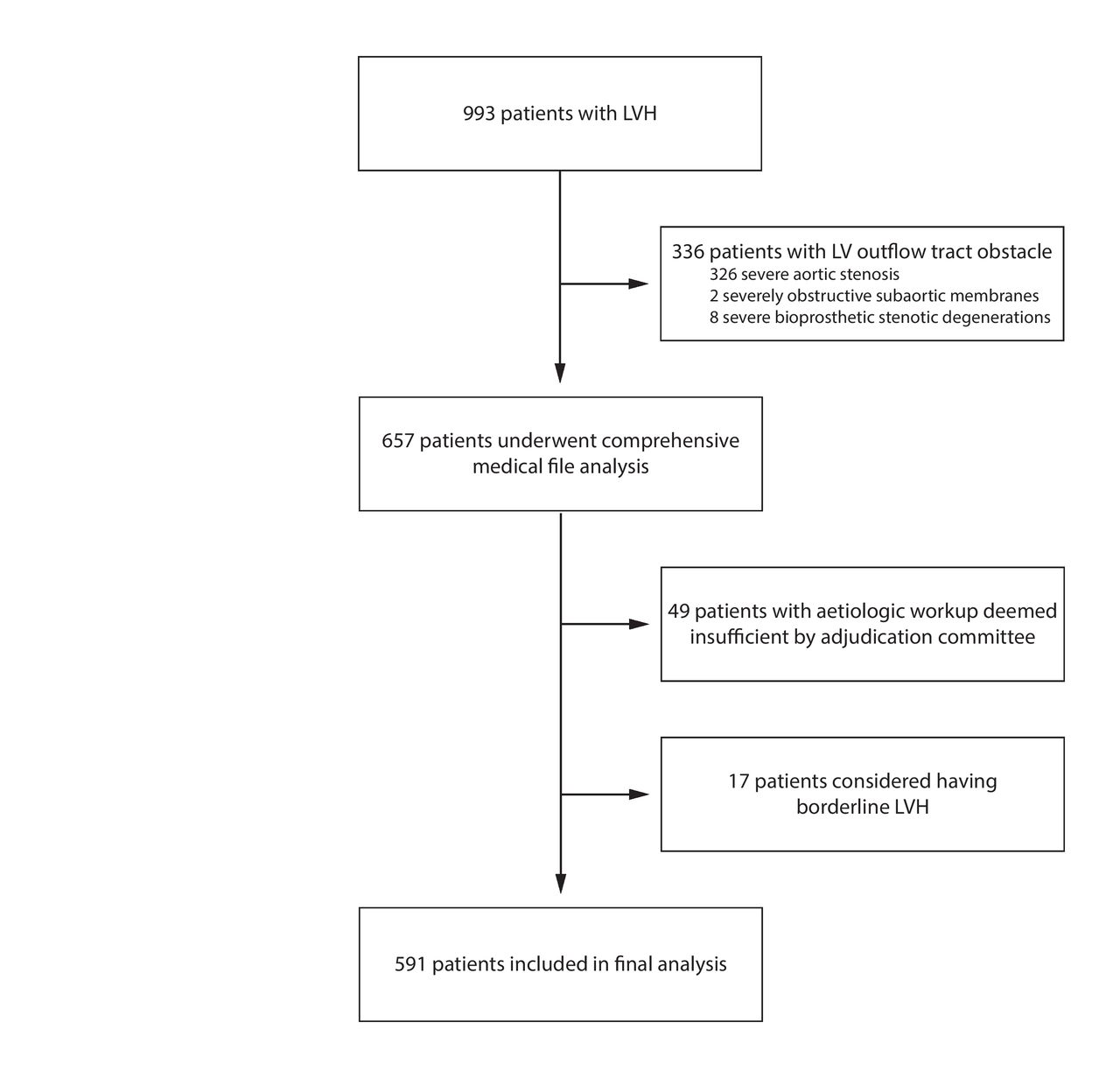
Nine hundred and ninety-three patients with LVH were admitted to our department of cardiac investigations during the studied period. All of them were referred by their cardiologist. Three hundred and twenty-six patients with severe AS were excluded, along with eight patients with bioprosthetic aortic valve stenotic degeneration and two patients with obstructive subaortic membrane. The adjudication committee considered the patient’s workup was lacking elements for 49 patients, withholding themselves to reasonably conclude on the LVH aetiology, which led to a posteriori exclusion. Finally, the concomitant occurrence of (1) borderline LVWT by TTE, (2) no LVH with CMR (maximum LVWT <12 mm) and (3) no aetiological lead after a complete workup led to conclude to a TTE false positive and to their withdrawal from the final analysis for 17 patients. The flow chart of the study is presented in figure 2.

Study flow chart. LV, left ventricle; LVH, left ventricular hypertrophy.
Population characteristics
Population characteristics are presented in table 1. Briefly, it was mainly composed of mildly symptomatic ageing men presenting with overweight. Arterial hypertension was notably prevalent as it affected 372 subjects (62.9%); it remained true whatever the LVH aetiology with a prevalence consistently over 50%. Electrical hypertrophy was common (132 patients, 22.4%) whereas history of ventricular arrhythmia or implantable cardioverter-defibrillator was rare (40 subjects, 6.8%). Population characteristics according to LVH aetiology are detailed in online supplemental material S1.
Supplemental material
Population characteristics
Main TTE findings are displayed in table 2. Concisely, the median of maximal LVWT was 15 mm, only 67.5% of patients had increased indexed LV mass, 69.7% had preserved LV ejection fraction (EF) but merely 16.5% had normal LV global longitudinal strain (≤−18%), and about 70% had normal right ventricular longitudinal function whether assessed by tricuspid annular plane systolic excursion or tricuspid annulus S’ wave velocity. Fourteen per cent of subjects presented with LV outflow tract obstruction with a median maximal gradient of 60 mm Hg. Strikingly, the most common aetiology in patients presenting with normal indexed LV mass was S-HCM (41.1%). Altered LV global longitudinal strain consistently reached about 75%–80% in each aetiology, rising up to 95% and 92% in cardiac amyloidosis (CA) and other aetiologies, respectively.
Transthoracic echocardiography findings
Main TTE findings according to LVH aetiology are detailed in online supplemental material S2.
Distribution of LV aetiologies
Distribution of LVH aetiologies is presented in online supplemental figure 1. Overall, the most common diagnosis was CA, found in almost a third of patients. The vast majority was transthyretin amyloidosis (ATTR) and especially wild-type ATTR (online supplemental material S3). Among patients with cardiac ATTR, 21.8% had inherited ATTR (aged 70 (64–73) years). Light-chain (AL) amyloidosis accounted for a fifth of CA cases. Anecdotally, three patients had cardiac involvement of AA amyloidosis and one of apolipoprotein A2 amyloidosis.
Supplemental material
Diagnosis of CA was closely followed by S-HCM, which affected more than a quarter of patients. Among those in whom genetic testing had been performed, most had no known mutation (53.0%; figure 3, online supplemental material S4); otherwise, most common mutated sarcomere genes were MYBPC3 (14.9%), MYH7 (11.2%), TNNT2 (1.5%), MYL2 (1.5%) and TNNI3 (0.7%) (genetic analysis was restricted to this set of genes in most patients).

{kind=link}
{kind=link}
{kind=link}
Genetic findings among patients with sarcomeric hypertrophic cardiomyopathy and known genetic status. MYBPC3, myosin binding protein 3; MYH7, myosin heavy chain 3; MYL2, myosin regulatory light chain 2; TNNI3, troponin I 3; TNNT2, troponin T 2.
HTN-CMP was the third main aetiology of LVH, it accounted for a fifth of the population.
Twenty-five (4.2%) patients had other aetiologies; mostly Anderson-Fabry’s disease (n=10, 1.7%), but also left ventricular non-compaction (LVNC) of the hypertrophic subtype (n=4, 0.7%), drug-induced HCM (tacrolimus n=2, 0.3%; hydroxychloroquine n=1, 0.2%), mitochondrial cytopathy (n=2, 0.3%) and isolated cases (0.1% each) of hypereosinophilic syndrome, Noonan syndrome with multiple lentigines, cirrhosis, transient myocardial oedema due to Tako-Tsubo syndrome, postcardiac arrest myocardial oedema and generalised lipodystrophy. LVH aetiology remained undetermined for 7.6% of the population.
Population below the European Society of Cardiology LVH threshold
Two hundred and forty (40.6%) patients had a maximum LVWT between 12 and 14 mm. Similar results were obtained using CMR measurement. Among them, 217 (90.4%) had a definite aetiology to their LVH.
Distribution of LVH aetiologies according to the LVH gradation is presented in table 3 (online supplemental figure 1). The distribution of LVH aetiologies significantly differed across the three grades of LVH (overall p<1.10−5; mild vs moderate p=0.16, moderate vs severe p<0.01, mild vs severe p<1.10−7). In this subset of the population, LVH aetiologies other than undetermined or HTN-CMP accounted for one half of subjects; undetermined aetiologies were not infrequent. Prevalence of CA and S-HCM increased with maximum LVWT whereas HTN-CMP, undetermined and other aetiologies decreased.
Left ventricular hypertrophy aetiologies detailed according to maximal left ventricular wall thickness
The three groups of LVH severity only differed on two clinical characteristics: age (higher LVH grades in older patients) and arterial hypertension (less frequent in higher LVH grades). These differences are driven by the increased prevalence of ATTR in more severe LVHs and of HTN-CMP in milder LVHs.
In the subset of patients presenting with CA, the distribution of each subtype of CA differed according to maximum LVWT. The proportion of AL-CA was greater in patients with milder LVH. In patients with ATTR, the proportion of inherited ATTR decreased with increasing maximum LVWT. Other forms of CA were only found in mild LVHs.
Considering subjects presenting with S-HCM, neither the prevalence nor the distribution of sarcomere gene mutations was influenced by LVH severity.
Discussion
In this study, we described the phenotype of patients referred to our tertiary centre for the aetiological work-up of LVH. The most common aetiology was CA followed by S-HCM and then HTN-CMP. LVH was only mild (LVWT 12–14 mm) in 41% of patients, yet meaningful diagnoses (in terms of prognosis, treatment and follow-up) could be made in more than half of them.
LVH definition
No reference supports the selection of 15 mm as the optimal cut-off for HCM diagnosis in the 2014 guidelines.1 Moreover, some room is left to perform a complete aetiological work-up for lesser degrees of wall thickening (13–14 mm), which suggests that they might need to be managed just the same as by the definition HCMs. Our study suggests a need in revising this definition: considering only patients with maximum LVWT ≥15 mm would have led to miss 146 (25%) LVH aetiologies other than HTN-CMP or undetermined. We are well aware that putting this into practice strictly would generate an important increase in healthcare expenses because ageing patients easily tend to present with maximum LVWT ≥12 mm. It may be useful to develop easily available ‘red flags’ (eg, young age, no or controlled arterial hypertension, family history, neuromuscular disorder, abnormal ECG, etc) that would trigger an aetiologic workup. We were confronted ourselves to that issue with some patients presenting borderline LVWT in TTE and a negative yield of the aetiological workup. In these patients, LVWT was not increased in CMR. We believe it is safe to consider them free of LVH (which does not necessarily mean free of heart disease). Maybe the use of the upmentioned red flags would have avoided further investigations.
An absolute cut-off of maximal LVWT to define LVH could in itself be criticised; such a boundary seldom applies indistinctly to all subgroups of a population. It may be interesting to adjust this cut-off to sex, height, race, LV afterload or physical activity (amount and type of exercise). Additionally, an absolute cut-off might overlook relative LVH in patients presenting with dilated cardiomyopathy.
HCM aetiologies
Our data suggest that the proportion of S-HCM may be overrepresented in European Society of Cardiology guidelines. A lead to explain this difference is that the studies they are based on have been focusing on a genetic standpoint, which may overlook non-genetic aetiologies, or even non-sarcomeric genetic aetiologies.
In our population, the genetic yield in patients with S-HCM is 36%. It is slightly below the range found by previous studies which extends from 38% to 53%,4–7 though fewer genes were analysed in most of our patients. Otherwise, we roughly found a similar distribution of the different mutated sarcomeric genes.5–8
A recently growing corpus of evidence suggests that prevalence of CA (especially wild-type ATTR) has been greatly underestimated in the past.9 10 Autopsy series showed 25% prevalence in the elderly.11 12 Almost 30% CA have been found diagnosed in patients with heart failure with preserved EF.13 Our study is in line with these recent findings; it is tempting to extrapolate that CA is at least just as prevalent as S-HCM, that is, 1 in 500 individuals. The up-mentioned underestimation of ATTR frequency also explains the high TTR-to-AL amyloidosis ratio we observed, unlike what have been published before.14
The exact proportion of inherited ATTR among patients presenting with cardiac ATTR is currently unknown. Two studies estimated this proportion at 12% and 36% of patients with cardiac ATTR.15 16 Accordingly, in patients who underwent genetic testing, we report 21.8% of TTR gene mutations, underlining the need of systematic genetic screening in patients with cardiac ATTR, regardless of age.
Genetic and enzymatic assay-based studies have estimated the prevalence of Anderson-Fabry’s disease from 0.5% to 3% in populations of HCM,8 17–20 which corresponds to this present study’s finding (1.5%).
Depending on definition and study population, LVNC prevalence varies from 0.01% to 3%.21 22 To our knowledge, no prevalence datum exists about the specific hypertrophic subtype of LVNC.
Though a linear correlation exists between the degrees of hypertension and LVH,23 24 the diagnosis of HTN-CMP remains challenging. The marked prevalence of hypertension across our population advises caution. Yet obviously necessary, history of arterial hypertension cannot be assumed sufficient to conclude to HTN-CMP, especially considering that some aetiologies of HCM (chiefly amyloidosis and Anderson-Fabry’s disease) generate renal impairment which can in turn induce hypertension.
Study limitations
Our study sharing all the limitations and biases associated with retrospective and single-site studies, our population does not reflect the entire panel of patients with LVH. Since the study was set in a tertiary care centre, our population is likely to select more complex cases than would have an ambulatory one, even more so with the exclusion of LV outflow obstructions. In this regard, HTN-CMP in particular may be underrepresented.
Additionally, we focused on patients who were hospitalised at least once in the cardiology wards of our hospital. This could explain why our study lacks neuromuscular disorder-associated HCMs (Friedereich’s ataxia, Pompe disease, Danon disease, etc) or other syndromic genetic HCMs (eg, Noonan syndrome), as their general follow-up is performed by other specialists and their cardiovascular evaluation seldom requires hospitalisation in a cardiology department.
Prospective multicentric studies are warranted to lift these limitations and define the most appropriate red flags to avoid unnecessary workups.
Conclusion
LVH begins before the arbitrary LVWT threshold of 15 mm and there is a wide range of diagnoses from the threshold of 12 mm. The evolution of prevention strategies and treatments according to the different aetiologies should encourage to review explorations thresholds to allow early detection and treatment.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors EC and OL participated in the design of the study. MB, EC, JB, AS, HD and SB were involved in data collection. MB performed the statistical analysis. MB, EC and OL interpreted the findings and drafted the manuscript and provided final approval of the version to be published. MB, EC, YL-B, DDD, MG, DC and OL contributed to the discussion. All authors have read and approved the final manuscript. OL is responsible for the final content as guarantor.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests Olivier Lairez received research support and personal compensation for consulting, serving on a scientific advisory board, speaking or other activities with Alnylam, Amicus, Genzyme, Pfizer and Shire-Takeda. Eve Cariou received personal compensation for speaking with Pfizer.
Patient consent for publication Not required.
Ethics approval Our institutional review board (CHU Rangueil) approved the study and registered it under the reference RnIPH 2020-85.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Data are available upon reasonable request to the corresponding author.