Article Text

Abstract
SARS-CoV-2 is the virus responsible for the ongoing COVID-19 outbreak. The virus uses ACE2 receptor for viral entry. ACE2 is part of the counter-regulatory renin-angiotensin-aldosterone system and is also expressed in the lower respiratory tract along the alveolar epithelium. There is, however, significant controversy regarding the role of ACE2 expression in COVID-19 pathogenesis. Some have argued that decreasing ACE2 expression would result in decreased susceptibility to the virus by decreasing available binding sites for SARS-CoV-2 and restricting viral entry into the cells. Others have argued that, like the pathogenesis of other viral pneumonias, including those stemming from previous severe acute respiratory syndrome (SARS) viruses, once SARS-CoV-2 binds to ACE2, it downregulates ACE2 expression. Lack of the favourable effects of ACE2 might exaggerate lung injury by a variety of mechanisms. In order to help address this controversy, we conducted a literature search and review of relevant preclinical and clinical publications pertaining to SARS-CoV-2, COVID-19, ACE2, viral pneumonia, SARS, acute respiratory distress syndrome and lung injury. Our review suggests, although controversial, that patients at increased susceptibility to COVID-19 complications may have reduced baseline ACE2, and by modulating ACE2 expression one can possibly improve COVID-19 outcomes. Herein, we elucidate why and how this potential mechanism might work.
- hypertension
- intensive care
- lung injury
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
I pictured myself as a virus or cancer cell and tried to sense what it would be like.—Jonas Salk
The counter-regulatory (also named alternative or non-canonical) renin-angiotensin-aldosterone system (RAAS), especially ACE2, the receptor for SARS-CoV-2, has been implicated in the pathogenesis of COVID-19.1 2 However, there is significant controversy whether overexpression of ACE2 would facilitate infection (given greater sites of entry) or restrict manifestations of the disease process (given favourable homeostatic effects).3–7 Due to its immediate relevance, we reviewed the available literature pertaining to SARS-CoV-2 interaction with ACE2, mechanistic significance of ACE2 signalling and specific drugs that could potentially modulate ACE2 expression. Referencing Dr Salk’s famous quote, to better understand potential therapeutic strategies against COVID-19, it would be important to either support or oppose repurposing commonly available drugs based on their impact on ACE2 expression or activity.
Basic virology
SARS-CoV-2 is an RNA virus of the same genus as the severe acute respiratory syndrome coronavirus of 2002–2003 (SARS-CoV) and the Middle East respiratory syndrome coronavirus (MERS-CoV).8 However, in terms of genetics and viral entry, SARS-CoV-2 more closely resembles that of other SARS viruses, including SARS-CoV, and uses the same receptor for entry into the host cells—ACE2; MERS uses dipeptidyl peptidase 4 (also known as CD26) receptors.9 10 This spike glycoprotein uses the serine protease TMPRSS2 for priming, essential for entry into ACE2 bearing target cells.2 ACE2 has 10-fold to 20-fold greater binding affinity to SARS-CoV-2 compared with SARS-CoV.1 11 SARS-CoV-2 expresses a unique coronavirus spike glycoprotein that does not cross-react with previously published SARS monoclonal antibodies.1
Tissue tropism
Since SARS-CoV-2 spikes the ACE2 receptor, it can be expected that cells with higher ACE2 expression would be the most vulnerable. With single-cell RNA-sequencing (scRNA-seq), ACE2 in humans is primarily expressed at higher levels in type II alveolar cells in the lung, proximal tubule cells in the nephron, myocardial cells, epithelial cells in the ileum and oesophagus, and urothelial cells in the bladder.12 However, nasal mucosa and bronchial samples, interestingly, showed less expression of ACE2. A separate study found high expression of ACE2 in the oral epithelium as well, especially along the tongue, although less than that seen in the lung, kidney and heart muscle.13 ACE2 receptors are more commonly expressed in the cells of the lower than the upper respiratory trees. In fact, there is evidence of persistent SARS-CoV-2 in sputum samples even days after symptoms have subsided.14 This probably explains why the most common COVID-19 signs and symptoms are lower respiratory in nature (ie, cough, expectoration, dyspnoea and lung infiltrates on imaging) without significant upper respiratory findings (ie, nasal congestion).15 16 Nonetheless, COVID-19 is highly contagious possibly due to the increased susceptibility of infection in the oral mucosa.15 16 Throat swabs from early on in the disease course found high viral isolation.14
It has also been theorised that the unique presence of a furin cleavage site along COVID-19’s glycoprotein could explain COVID-19’s ability to bind to tissue with relatively lower ACE2 expression.17 Since furin is a widely expressed protease in the respiratory tract, COVID-19 uses it to increase its infectiveness and transmissibility.17 The furin site is not present in other SARS viruses, but is found in certain avian influenza viruses and MERS-CoV.18–20 Both avian influenza and MERS-CoV viruses were significantly less infectious compared with COVID-19; thus, a combination of other viral characteristics is most likely to blame for COVID-19’s high infectious rate. An in vitro study proposed that COVID-19’s S protein was able to undergo syncytium formation with ACE2+ cells in a protease-independent fashion, thereby promoting viral spread.21 However, the exact significance and efficiency of COVID-19’s protease-independent syncytial formation in vivo require further investigation. A recent work showed that both cleavage and syncytium were abolished by treatment with furin inhibitors decanoyl-RVKR-chloromethylketone and naphthofluorescein. These furin inhibitors might become promising antiviral agents for prevention and treatment of SARS-CoV-2 infections.22
ACE2 binding
The efficiency of SARS strains to bind to ACE2 may be an indicator of their infectiveness in humans. This is extrapolated from the observation that SARS-CoV more effectively used ACE2 compared with the SARS strain of the lesser outbreak in 2003–2004. Similarly, the SARS coronavirus strains found on civets neither effectively target ACE2 and therefore did not infect humans.23 Computer modelling confirms that although SARS-CoV-2 has higher affinity to ACE2, its receptor-binding domain is not as efficient in recognising ACE2 as SARS-CoV; however, SARS-CoV-2 is more efficient in recognising the ACE2 receptor compared with the SARS strain of the smaller outbreak in 2003–2004.10 24
Cytotoxicity
Although the cytotoxic strength of SARS-CoV-2 following ACE2 binding is yet to be compared with that seen with SARS-CoV, the more potent cytotoxic potential of SARS-CoV-2 might be due to its greater efficiency in using ACE2 receptors. In addition to the increased travel volume over the past two decades, the differences between SARS-CoV-2 and SARS-CoV affinity to the ACE2 receptors most likely explain why the SARS-CoV-2 outbreak has become far more widespread than the SARS-CoV outbreak. SARS-CoV-2 has sufficient binding capacity to ACE2 for human-to-human transmission yet is far more likely to cause no or mild symptoms in most cases. This makes it extraordinarily difficult to identify cases and to initiate quarantine.
Pathogenesis
Lung injury
The actual mechanism of COVID-19 manifestation after ACE2 receptor spiking remains to be elucidated. However, since SARS-CoV-2 shares similar structural features with SARS-CoV, we can theoretically extrapolate its pathogenesis from previous knowledge. In vivo models show that SARS-CoV spike protein primarily localises to ACE2 receptors on bronchial epithelial cells and alveolar pneumocytes.25 SARS-CoV spike protein significantly enhanced the severity of lung injury following chemically induced inhalation injury; however, the presence of spike protein alone was not sufficient to induce significant lung disease.25 The virus then gains entry to the cell by endocytosis, resulting in downregulation of ACE2 protein expression in the lungs. This is due to activation of A disintegrin and metalloproteinase-17 (ADAM17) cleaving the ACE2 N-terminal.25 26 Therefore, we could surmise that SARS-CoV-2 also decreases ACE2 expression (figure 1).10 24
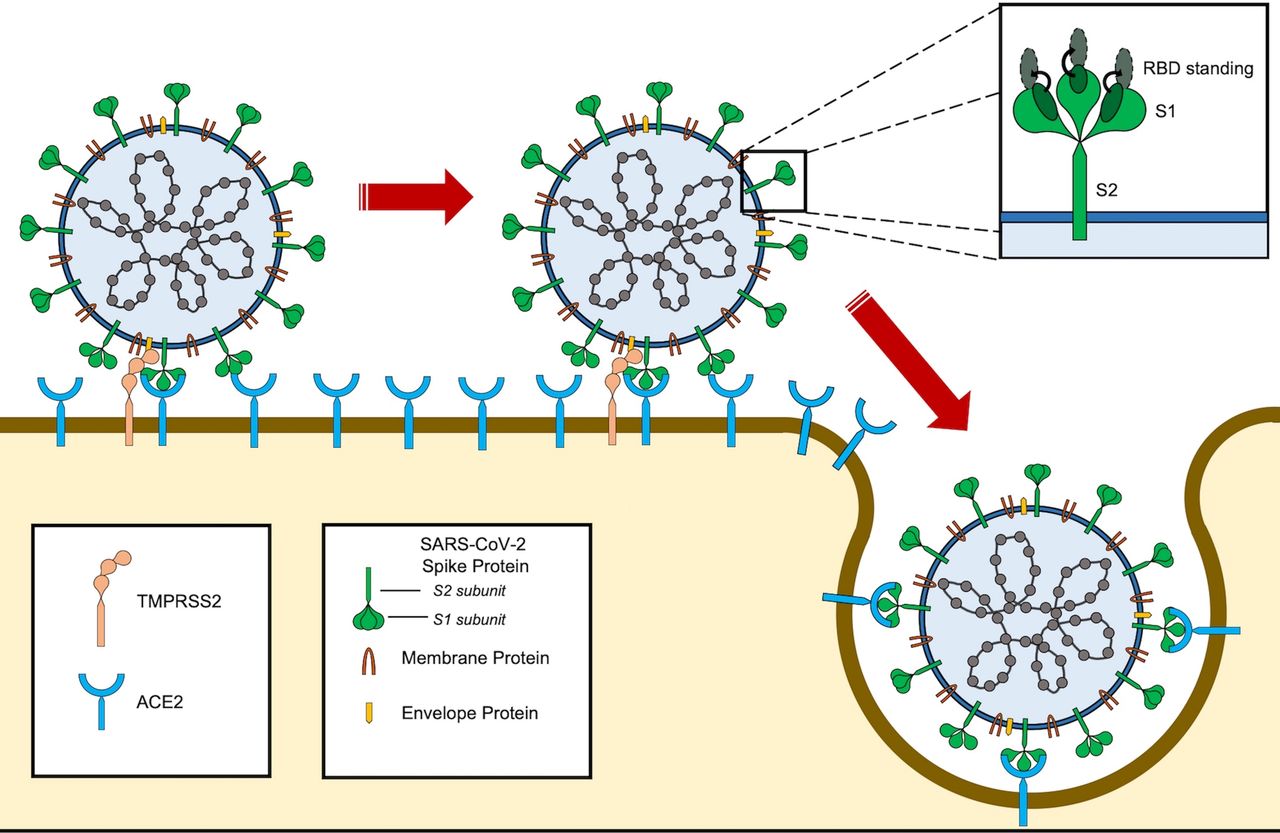
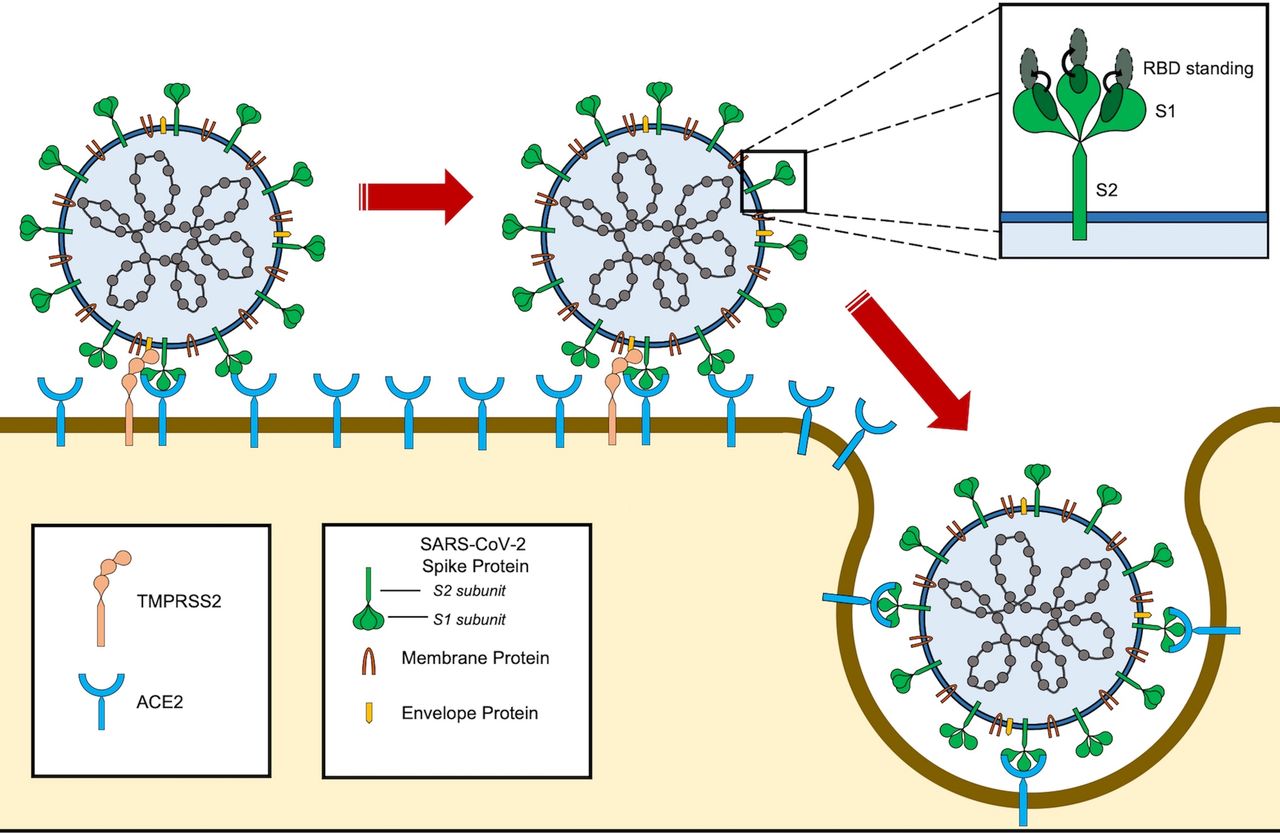
SARS-CoV-2 entry into the cell. SARS-CoV-2 binds to ACE2 through the receptor-binding domain (RBD) of spike protein. The L455, F486, Q493, S494, N501 and Y505 residues in SARS-CoV-2 RBD interact with K31, E35, D38, M82 and K353 residues of ACE2 receptor. Notably, the RBD of SARS-CoV-2 tends to generate a large binding interface of 1204 Å, along with an extended contact with the N-terminal helix of ACE2, and the higher affinity of the virus for ACE2 binding. For entry, S1 and S2 subunits dissociate from each other through cleavage by a protease, such as TMPRSS2. Furin also cleaves the site between both S1 and S2 subunits of the SARS-CoV-2 spike protein and serves as the alternative pathway of activation, especially in cells with lower TMPRSS2 expression.108
In the classic RAAS cascade, ACE converts angiotensin I (Ang I) to angiotensin II (Ang II). Ang II then activates Ang II type 1 receptor (AT1R), classically leading to vasoconstriction, but also promotion of inflammation, fibrosis and microvascular permeability.27 ACE2, an ACE homologue, is a part of the vasoprotective/anti-inflammatory axis of the counter-regulatory RAAS, which also includes Ang II type 2 receptor and the angiotensin-(1–7) (Ang-(1–7))-mitochondrial assembly (MAS) receptor axis.28 29 ACE2 naturally converts Ang I to angiotensin-(1–9), which is then converted to Ang-(1–7) by ACE or by neutral endopeptidase.29 ACE2 also converts Ang II directly to Ang-(1–7); the latter pathway to creating Ang-(1–7) has significantly higher catalytic efficiency.28 Ang-(1–7) then goes on to activate the MAS pathway.
Decreased ACE2 results in increased Ang II availability that could bind to AT1R. This should increase vascular permeability, resulting in non-cardiogenic pulmonary oedema and severe acute lung injury.27 In a small study of 12 patients with COVID-19 in Shenzhen, China, there was a linear association between viral load and lung injury with Ang II, but was not found to be statistically significant (p=0.06), likely due to inadequate study power.30 However, it should be of note that the role of pulmonary oedema in COVID-19 remains unclear. A common clinical presentation of non-cardiogenic pulmonary oedema is dyspnoea with hypoxaemia and an absence of cardiogenic aetiology (ie, low pulmonary arterial wedge pressures). Interestingly, it was shown by Guan et al15 that in hospitalised patients with COVID-19, dyspnoea is relatively low (18.7%) despite a high prevalence of hypoxaemia, abnormal CT scans and requirements for supplemental oxygen. This phenomenon has been termed as ‘happy hypoxaemia’.31 More studies are required to further evaluate this unique presentation of pulmonary oedema in COVID-19.
Increased Ang II in the plasma has been shown to correlate with the severity of lung injury for other viral infections as well. In mice infected by respiratory syncytial virus (RSV), ACE2 expression decreased in the lung tissue and was correlated with an increase in plasma Ang II levels and lung injury severity.32 Furthermore, ACE2 knockout mice suffered from more significant lung injury compared with wildtype mice.32 Plasma Ang II levels were shown to be strongly associated with mortality in patients who suffered from influenza A/H7N9 (also known as avian influenza virus) during the outbreak in China in 2013.33 In fact, the model strength of using Ang II to predict mortality was higher than that of C reactive protein (area under the curve, AUC=0.875 vs AUC=0.76) and various respiratory functional metrics, including partial pressure of oxygen (PaO2)/fraction of inspired oxygen (FiO2) (AUC=0.75) and Pneumonia Severity Index score (AUC=0.825).33 However, ACE2 expression was not obtained. Nonetheless, we can speculate that alterations in the RAAS resulting in decreased ACE2 and increased Ang II aggregate lung injury from certain viral infections.
Inflammatory response
The decreased expression of ACE2 prevents the degradation of des-Arg9 bradykinin (DABK) by the epithelial cells in the lungs.34 Increased DABK results in the activation of DABK/bradykinin receptor B1, which in turn results in the release of proinflammatory chemokines by the lung epithelium. Specifically, C-X-C motif chemokine ligand 5 (CXCL5), macrophage inflammatory protein 2 (MIP2), C-X-C motif ligand 1 (CXCL1) and tumour necrosis factor-alpha (TNF)-α are released, leading to an exaggerated neutrophilic response, thereby worsening lung injury. Additionally, overexpression of ACE2 via genetic therapy increased the production of Ang-(1–7), upregulation of the MAS pathway, inhibition of the extracellular signal-regulated kinase (ERK)/nuclear factor kappa B (NF-κB) pathway, and hence prevention of exaggerated inflammatory response seen in acute respiratory distress syndrome (ARDS) (figure 2).35 36 Decreased ERK/NF-κB activation via MAS has been shown to reduce transforming growth factor-beta (TGF-β) and interleukin (IL)-6.36 If SARS-CoV-2 also decreases ACE2 expression, we would expect that there would be less Ang-(1–7) production. This would decrease activation of MAS, leading to an upregulation of ERK/NF-κB, as also TGF-β and IL-6 release to significantly increase inflammatory and fibrotic responses. Interestingly, Peiró and Moncada37 recently proposed that the use of Ang-(1–7) or one of its mimetics should be considered among other strategies to prevent damage in high-risk patients with COVID-19.
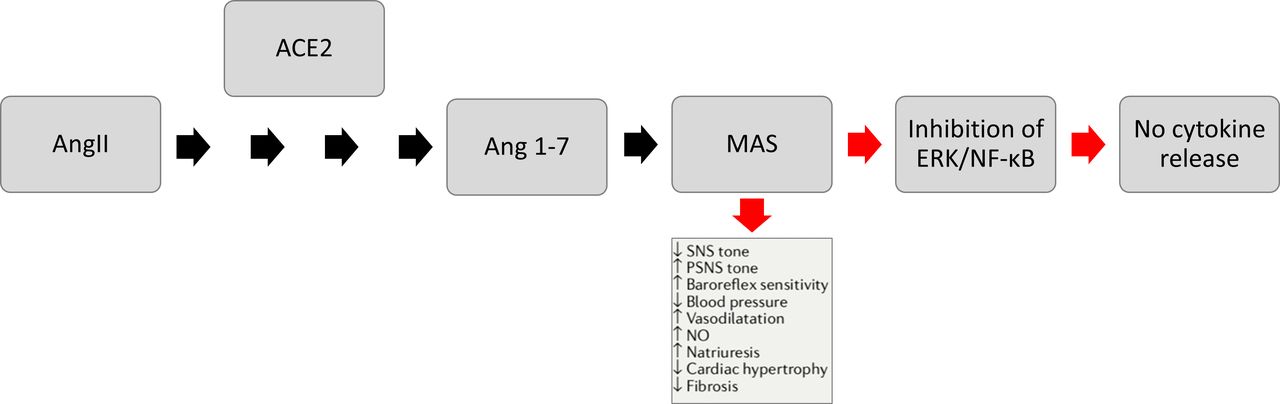
The ACE2 mechanism. Ang II is converted by ACE2 to become Ang-(1–7). Ang-(1–7) activates the MAS receptor signalling pathway, which then inhibits ERK/NF-κB, resulting in no cytokine release. Ang II, angiotensin II; ERK, extracellular signal-regulated kinase; MAS, mitochondrial assembly; NF-κB, nuclear factor kappa B; NO, nitric oxide; PSNS, parasympathetic nervous system; SNS, sympathetic nervous system.
Indeed, several early studies show that COVID-19 severity is associated with increased plasma levels of cytokines such as IL-6.38–40 Furthermore, in a meta-analysis, procalcitonin level, which is a known marker of systemic inflammatory response syndrome, was found to be fivefold higher in patients with severe COVID-19.41 42 Therefore, procalcitonin and IL-6 measurements provide a window to the assessment of severity of COVID-19 and improved prediction for prognosis.
A recent transcriptional analysis of bronchial alveolar lavage (BAL) samples and peripheral blood mononuclear cells (PBMCs) from three patients with COVID-19 demonstrated significantly higher expression of proinflammatory cytokines, including TGF-β, in both samples, compared with that seen from three healthy donors.43 Interestingly, there was little detection of viral RNA in PBMCs, yet there was significant activation of proinflammatory pathways (including neutrophilic activators) and apoptotic in lymphocytes. The virus, therefore, can induce apoptosis in lymphocytes without direct infection. They also did not find increased expression of IL-6 in PBMCs or IL-6 receptors in the lung epithelium, thus possibly contradicting previous findings showing increased IL-6 in blood correlating with disease severity. However, given that there is no clinical information regarding severity or treatment of these three patients, it is difficult to identify IL-6’s role in the disease. Furthermore, the authors stated that lymphopaenia in infected patients could be attributed to either endogenous or exogenous corticosteroids. Nonetheless, evidence points towards excessive cytokine release as a fundamental aspect towards COVID-19 pathogenesis. Thus, as expected, suppressing this inflammatory response with dexamethasone has shown to reduce mortality in severely ill patients with COVID-19.44
Acute respiratory distress syndrome
Initial physician reports of COVID-19 indicated that ARDS was not a commonly reported diagnosis during hospital admission (3.4%) and that most of the patients received a diagnosis of pneumonia (91.1%).15 However, it was identified via a meta-analysis of 10 early COVID-19 papers that there were significant variation and significant publication bias in regard to reporting the incidence of ARDS (p=0.008 via Egger’s test).16 Therefore, it is difficult to ascertain whether these initial studies reliably reported the incidence of ARDS.
ARDS is often difficult to distinguish from pneumonia without BAL because significant bilateral pneumonia clinically mimics ARDS.45 Using an unbiased standardised measurement for assessing and reporting the incidence of ARDS could help reduce publication bias. A single-centre retrospective study reported that of the 201 initial patients who were hospitalised with confirmed COVID-19 pneumonia, 84 (41.8%) developed ARDS as defined by the WHO Interim Guidelines (based on the Berlin definition), of which 44 died (52.2%).46 47 Similarly, a multicentre retrospective study that also used the Berlin criteria for ARDS found that of the 191 patients hospitalised with COVID-19, 59 (31%) developed ARDS (the median time from onset of illness to ARDS was 12 days), resulting in more of these patients dying (93%).48 Severely ill patients may develop dyspnoea and hypoxaemia within a week of symptom onset. The development of ARDS was most strongly associated with older age, although interestingly neutrophilia was also found to be associated with ARDS, although modestly.
The first postmortem case reports of biopsies of lungs from patients with COVID-19 demonstrated significant bilateral diffuse alveolar damage with cellular fibromyxoid exudates.49 50 There were signs of significant desquamation of the pneumocytes, hyaline membrane formation and pulmonary oedema classically seen with ARDS. There was evidence of intense interstitial mononuclear inflammatory infiltrates which were dominated by lymphocytes. There was also evidence of enlarged amphophilic granular pneumocytes indicating viral cytopathic-like changes. The lung biopsy indicated significant inflammatory response and oedema, like that seen in downregulated ACE2 animal models and similar to that seen from SARS-infected animals and from SARS-infected human lung biopsies.25 51 This finding also correlates with that seen from postmortem lung samples infected with H5N1, showing significant lung injury due to an exaggerated inflammatory response via excessive cytokine release.52
Possible postinfection consequences
There are few data regarding patient outcomes after resolution of COVID-19 pneumonia due to its novelty. However, it can be reasonably inferred that some patients with significant COVID-19 pneumonia and lung damage will subsequently develop chronic fibrotic change in the lungs. Patients with viral pneumonia are usually at increased risk of pulmonary fibrosis, possibly due to release of TGF-β to promote tissue repair after significant inflammatory response.53 Two studies of surviving patients with SARS 2002 pneumonia showed significant fibrotic changes present in 45% of survivors at 1 month in one of the studies, while the other study demonstrated significant lung fibrosis in 36% of survivors at 3 months and 30% at 6 months.54 55 Additionally, exercise capacity and quality of life of these patients were compromised compared with the normal population. Therefore, patients who have survived significant COVID-19 pneumonia would need to be monitored for potential respiratory deterioration and fibrotic consequences.
Risk factors for COVID-19 complications and ACE2 expression
Age and sex
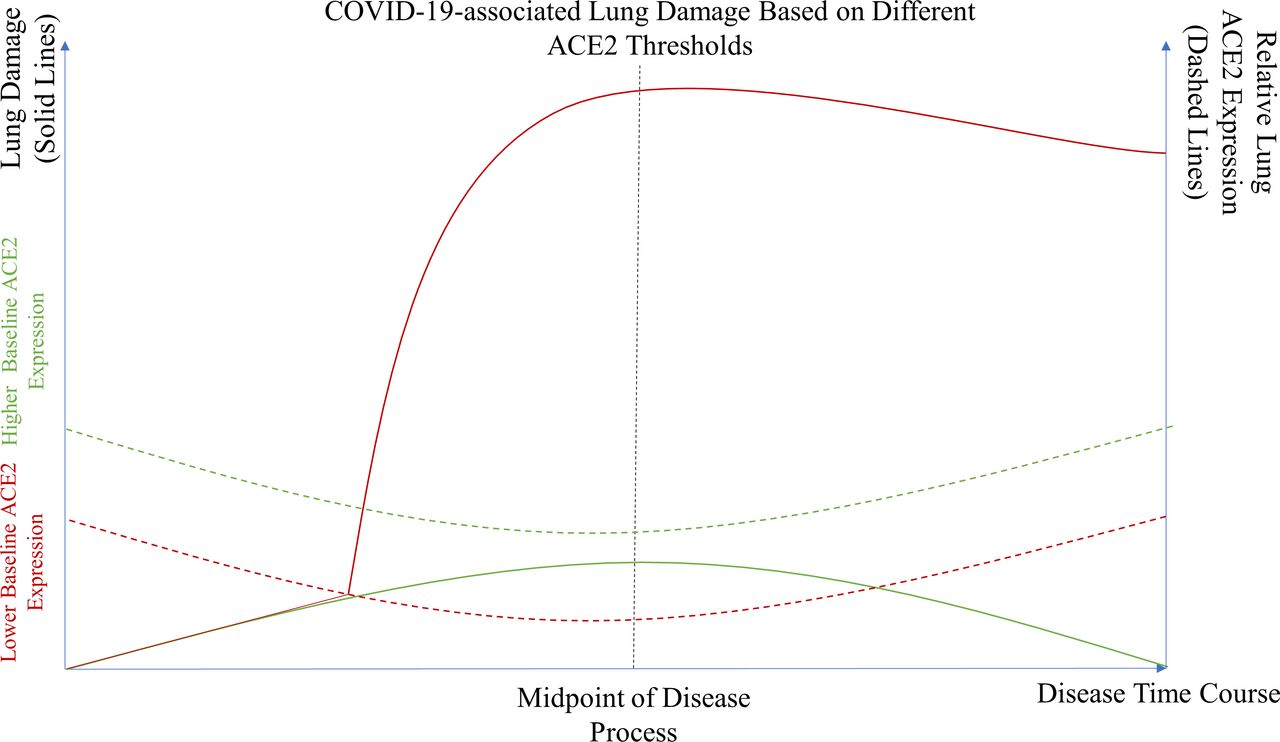
As discussed earlier, decrease in ACE2 expression levels beyond a certain threshold by COVID-19 is the most likely explanation for progression to severe lung damage and potential fibrosis (figure 3). Therefore, prevention of ACE2 from reaching below this threshold by increasing its relative expression would most likely help prevent worsening lung injury.

SARS-CoV-2-associated lung damage based on different ACE2 thresholds. A theoretical model illustrating the severity of lung damage in patients with normal or higher baseline expression (green solid line) versus those who have lower baseline ACE2 expression due to certain comorbidities or risk factors (red solid line). Patients with higher baseline ACE2 expression never meet their critical ACE2 expression threshold (green dotted line) and thus are less likely to experience significant lung damage. Meanwhile, patients with lower baseline ACE2 expression are more likely to reach their ACE2 expression threshold (red dotted line) and therefore suffer more severe lung injury.
It has been shown that ACE2 expression in animal models decreases with ageing and is lower in men than in women (oestrogen upregulates ACE2; also ACE2 is on the X chromosome), thereby explaining the phenomenon that older men are more likely to succumb to COVID-19-associated pulmonary complications.56 57 In human atrial myocardium samples, 17β-oestradiol expression (E2) significantly increased the expression of ACE2.58 After menopause the E2 levels decrease and therefore ACE2 expression could also decrease; however, it is not known if this is significant enough to reduce its protective effect in the lungs.59 Nonetheless, age in and of itself is a strong risk factor for COVID-19-associated complications. Compared with COVID-19-infected patients aged 30–59, infected patients below 30 were less about half as likely to die, while infected patients over 59 were at a fivefold increased risk of dying.60 Furthermore, the risk of symptomatic infection increased with age at about 4% per year for patients between 30 and 60 years old.60
Hypertension
Hypertension has been strongly associated with COVID-19 pneumonia and is one of the most commonly associated comorbidities seen in patients with severe COVID-19 pneumonia.46 61 In a preprint study, Lu et al62 analysed the characteristics of 265 patients with COVID-19 and found that 17.2% of mild to moderate patients were hypertensive and 45.5% of severe patients were hypertensive. This suggests that patients with COVID-19 who were hypertensive may correspond to more severe cases. Other initial reports demonstrated similar findings.63 64 A multicentre retrospective study of 191 COVID-19 hospitalisations found that hypertension was significantly associated with non-survivors (48% vs 23%, p=0.0008) and was a significant risk factor for in-hospital death (OR, 3.05; 95% CI 1.57 to 5.92).48 Overall, a recent meta-analysis of 10 articles reporting 3403 patients in total found that hypertension was the most prevalent underlying comorbidity in hospitalised patients with COVID-19.65 However, this may be caused by higher portions of hospitalised patients being older and are therefore more prone to being hypertensive.
It is known that hypertension decreases the expression of ACE2 significantly; thus, patients with hypertension are more prone to have ACE2 levels below a certain threshold, making them more vulnerable to significant lung pathology from COVID-19 infection.66 ACE2 serves as a negative regulator of RAAS and can directly metabolise Ang II to generate the beneficial heptapeptide Ang-(1–7), whose actions are often opposite to those attributed to the Ang II and its type 1 receptor (AT1R) signalling. A recent study suggested that in patients with hypertension and comorbid heart failure, the plasma ACE2 levels decreased and were accompanied by upregulation of Ang II levels and downregulation of Ang-(1–7) levels.67 After treatment with recombinant human ACE2 in these patients, plasma Ang II was downregulated and Ang-(1–7) level increased, suggesting that ACE2 regulates Ang II metabolism in humans.
Smoking and pollution
Normally, one would expect that patients who smoke would be at higher risk for lung complications associated with viral infection.68 However, smoking has failed to show a significantly high association with severity of COVID-19 pneumonia.46 48 63 69 70 In the same study mentioned above, of the 191 COVID-19 hospitalisations, there were no differences in smoking prevalence between non-survivors and survivors (9% vs 4%, p=0.21) and smoking was not a significant risk factor for in-hospital death (OR, 2.23; 95% CI 0.65 to 7.63).48 Nonetheless, it did appear to trend towards an association. Interestingly, according to a preprint scRNA-seq study, Asian smokers and former smokers had significantly higher ACE2 expression in the lungs compared with that seen with non-Asian smokers, although Caucasian smokers did not have higher ACE2 levels.71 This preliminary study may indicate a gene–smoking interaction conferring to varying ACE2 levels depending on genes and smoking. Brake et al72 demonstrated visually in surgically resected lung tissue that there were more ACE2 receptors in patients who smoke with normal lung function and those who have chronic obstructive pulmonary disease (COPD) when compared with non-smokers. Regardless, smoking does increase one’s risk for hypertension, cardiovascular disease and COPD, which were all associated with increased risk for severe COVID-19 pneumonia in initial Chinese studies.48 73 Therefore, we expect that smoking would eventually be a significant risk factor once larger more diverse studies are published.
Recently, however, a preprint study identified that long-term exposure to fine particulate matter (PM2.5) from air pollution may be associated with increased mortality rates in patients with COVID-19.74 It has been shown that PM2.5 leads to significant pulmonary inflammation by inducing RAAS.75 76 In a mouse model, ACE2 deficiency resulted in slower lung recovery from PM2.5.77 This indicates that ACE2 may be a protective responder to chronic particle exposure, although COVID-19 may overburden ACE2’s protective capability by further exacerbating the underlying inflammatory process.
Chronic obstructive pulmonary disease
One study identified, via a meta-analysis of available transcriptomic lung data sets, that ACE2 is expressed higher in patients with COPD compared with controls.78 The previously mentioned study by Brake et al also found more staining of ACE2+ cells in the lung tissue.72 COPD does in fact have a strong association with worse cases of COVID-19 pneumonia. One larger retrospective study of 1590 patients with COVID-19 from China showed that COPD, after adjusting for age and smoking status, was strongly associated with adverse composite endpoint, defined as admission to intensive care unit (ICU), invasive ventilation or death (HR, 2.68; 95% CI 1.42 to 5.048).79 Another study of 339 patients with COVID-19 who were over 60 from Wuhan, China found that COPD was more prevalent in patients who died (HR, 3.72; CI 1.94 to 7.13, p<0.001).80 Overall, though, there has not been strong evidence correlating COPD with increased infectious risk with COVID-19.65 This may indicate that the virus is still highly infectious regardless of ACE2 expression, and the increased risk of poor outcomes could stem from poorer lung function rather than increased ACE2 expression.
Differences between circulating ACE2 and membrane ACE2
Circulating ACE2 (soluble ACE or sACE2) is produced after being shed from cell membranes due to a disintegrin and a metalloprotease ADAM17; therefore, membrane ACE2 and plasma ACE2 activities and expressions do not necessarily positively correlate with one another.81 Plasma ACE2 is significantly less catalytic and less abundant than membrane ACE2 found in the lung epithelium. In fact, ACE2 plasma expression is essentially undetectable in healthy individuals, about 100-fold lower in expression compared with ACE plasma expression. Nonetheless, increased ACE2 in plasma correlates highly with cardiovascular disease risk factors.82 In the Leeds Family Study, out of 534 healthy white Europeans from 89 pedigrees of 133 households, only 40 had noticeably detectable ACE2 plasma expression above 2.7 pM.82 These 40 individuals were older, had higher waist/hip ratios, higher rates of hypertension, elevated fasting glucose, hypertriglyceridaemia, hypercholesterolaemia and elevated neprilysin levels. High-density lipoprotein levels were the only statistically significant laboratory value that was positively correlated with quantitative circulating ACE2 levels (p=0.03).
Circulating ACE2 has also been found to be mostly expressed in patients with heart failure, and expression was correlated with severity of heart failure.83 In circulating ACE2 67% of variance was ascribed to genetic factors and had marked familial clustering.82 Therefore, using circulating ACE2 expression as a marker of ACE2 activity would be difficult for use in the general population. Contrarily, detectable expression levels may be more likely found in patients with decreased ACE2 expression in the lungs due to excessive shedding from cellular membranes by ADAM17.81
Potential therapeutic interventions for manipulation of ACE2
Ang II receptor blockers
ACE2 receptor works in concert with AT1R. When Ang II binds to AT1R, the ACE2 receptor dissociates from AT1R and it is internalised and degraded intracellularly (figure 4).84 Additionally, ACE2 competes with AT1R for local Ang II. Thus, it has been shown that Ang II receptor blockers (ARBs) significantly increase ACE2 receptor expression in animal models by preventing its dissociation.85 ARBs therefore in COVID-19 could contribute to (1) preventing the reduction of ACE2 to a certain threshold that leads to overt cytokine release and (2) preventing pulmonary oedema and acute lung injury due to overactivation of AT1R in the presence of Ang II.27
{kind=link}
{kind=link}
{kind=link}
{kind=link}
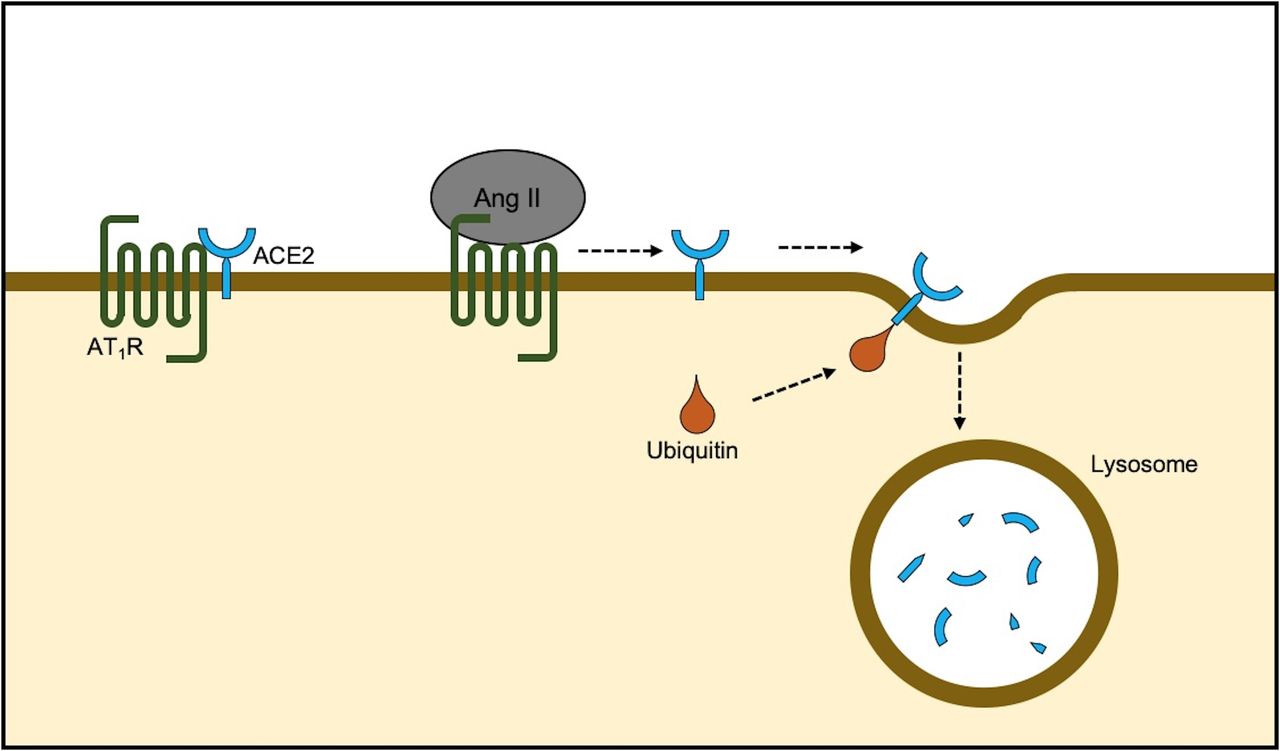
Natural degradation of ACE2. ACE2 remains in approximation of AT1R on the cell membrane. Arrival of Ang II decreases the physical association and results in ubiquitination and ACE2 internalisation followed by lysosomal degradation. AT1R antagonism prevents degradation by stabilisation of the ACE2-AT1R complex and also renders favourable effects by mitochondrial assembly receptor activity.84 Ang II, angiotensin II; AT1R, Ang II type 1 receptor.
In humans, however, the use of ARBs has not been shown to significantly increase plasma ACE2 levels, causing scepticism about the potential role of ARBs on ACE2 potentiation in humans. However, as mentioned, it would be difficult to detect noticeable differences in ACE2 expression and activity solely based on plasma samples. Therefore, measuring plasma ACE2 protein levels is a poor option to assess ACE2 activity after therapy. Up-to-date clinical evidence for ARBs to treat COVID-19 is unavailable. Regardless, in a retrospective study of 22 996 patients, inpatient ARB use for hospitalised patients with pneumonia over the age of 65 was associated with reduced 30-day mortality (OR, 0.47; 95% CI 0.30 to 0.72).86 Also, in patients with COPD, ARBs were reported to be associated with reducing the risk of pneumonia.84 87 Thus, even though COPD is associated with increased ACE2 expression, it is possible that increasing ACE2 expression even further with ARBs could still be lung-protective. Furthermore, Soto et al88 conducted a respective cohort including 215 225 patients and found there is an association between the use of ARBs and a delay in the progression of pulmonary complications.
Similarly, in an experimental setting where lung injury was induced by SARS spike protein and acid challenge, losartan therapy was associated with substantially attenuated damage; therapeutic benefits were almost immediately appreciable with significant change in per cent elastance.25 Since ARBs are low-risk, inexpensive and widely available drugs, it would be of considerable interest to see if ARBs could attenuate or even prevent significant lung injury in patients with COVID-19. Given the benefits, ARBs would be most beneficial in the early disease process to prevent lung injury. Therefore, it is important to establish retrospective cohort studies or prospective randomised controlled trials to observe the effect of ARBs on the clinical outcome of COVID-19. Fortunately, trials are currently under way to investigate the effects of ARBs on COVID-19-related complications (NCT04312009, NCT04311177).
ACE inhibitors
ACE inhibitors (ACEis) also increase ACE2 levels by reducing the conversion of Ang I to Ang II, thereby preventing Ang II binding to AT1R and ACE2 internalisation.85 ACEis were also shown to be significantly associated with reduced 30-day mortality in the previously mentioned study of hospitalised patients with pneumonia. ACEi in a murine model protected against acute lung injury after lipopolysaccharide inhalation by increasing ACE2 expression and decreasing IL-6 and TNF-α release36; however, this protective effect and its effect on ACE2 is controversial.3 4 In a murine myocardial ischaemia-reperfusion model, enalaprilat administration was associated with inhibition of kininase II activity and in increased DABK formation, which could promote an inflammatory response.89 Given the more direct effect of ARBs to enhance ACE2 expression, ARB might comprise a better initial candidate than ACEis.
Other RAAS antagonists
Mineralocorticoid receptor antagonists (MRAs) have shown to induce ACE2 expression in murine macrophages and the heart after 1 month of therapy.90 MRA also restricted neutrophil and macrophage infiltration in the lungs after bleomycin exposure and attenuated cytokine release of IL-6 and TNF-α.91 Interestingly, aldosterone itself failed to affect ACE2 expression and activity in human mesangial cell in vitro, but increased ACE activity and decreased MAS receptor expression.92 The reduced MAS might promote an inflammatory response, and the increased ACE activity could increase in Ang II systemically, leading to further reduction in ACE2 expression. In the mesangial cells in vitro, Ang II most likely does not bind to AT1R in the presence of MRA. Intranuclear sequestration of Ang II by MRA reduces its binding to AT1R systemically resulting in increased ACE2 expression. On the contrary, spironolactone treatment decreased MAS receptor levels by reducing local Ang-(1–7) expression in mesangial cells, thereby increasing DABK activation and cytokine release. However, given that MRA systemically appears to reduce Ang II expression, it may be protective in the lungs, although more studies are required. Regardless, like ACEi, there is less direct evidence that MRA produces similar protective effects in a viral-related acute lung injury.
Contrary to other RAAS inhibitors mentioned, the renin antagonist aliskiren was associated with reduced ACE2 expression in a diabetic nephropathy rat model after 6 weeks of treatment.93 The mechanism of this has not yet been explored, although we expect increasing ACE2 to be more lung-protective compared with decreasing ACE2. Therefore, aliskiren would not be a desirable therapeutic option.
Diminazene aceturate
Diminazene aceturate (DIZE) is a Food and Drug Administration-approved anti-trypanosomal medication that was identified via computational modelling to activate ACE2; it was then shown to be cardioprotective against myocardial infarction and could attenuate pulmonary hypertension by increasing ACE2 activity in preclinical models.94 95 DIZE has been shown to reduce the severity of inflammation and oxidative stress from hyperoxic lung injury, most likely by increasing ACE2 activity by promoting MAS inhibition of DABK.96 DIZE has yet to be studied in the setting of viral pneumonia; however, given its ability to increase ACE2 expression and to attenuate lung injury, it could be an interesting experimental therapeutic to protect the lung from certain ACE2 modulating viral infections.
Soluble ACE2
ACE2 soluble proteins have been in development for several years now to treat ARDS. A recombinant form of human ACE2 (GSK2586881) was under clinical investigation to attenuate lung injury associated with ARDS and was shown to be safe and effective in decreasing Ang II in humans.97 Furthermore, it was shown that sACE2 attenuated lung injury in mice after infection from RSV and exposure to SARS spike protein.27 32 Recently, it was demonstrated that sACE2 prevented the entry of SARS-CoV-2 into cell lines by saturating the virus binding sites.98 Therefore, it has been theorised that sACE2 both could reduce lung injury associated with decreased ACE2 and could also potentially prevent viral entry.99 Although this molecule is currently not available for clinical use, this could be a promising therapeutic option to reduce SARS-CoV-2 viral load, possibly reducing COVID-19-associated complications. There are currently expedited clinical trials to evaluate if intravenously administered sACE2 would be effective in treating COVID-19 illness (NCT04287686).
Statin therapy
As mentioned before, COVID-19 mortality is strongly associated with history of cardiovascular disease. Dyslipidaemia leading to atherosclerotic plaque formation is a main risk factor resulting in cardiovascular disease. In a rabbit model, it was shown that high-cholesterol diet resulted in downregulation of ACE2 protein expression in the heart and kidneys, but interestingly not mRNA activity.100 Treatment with atorvastatin increased histone H3 acetylation of the ACE2 promoter region in the heart, resulting in increased ACE2 mRNA expression, yet not the kidney. Atorvastatin did, however, increase ACE2 protein expression overall in the kidneys and in the heart. The disconnection between ACE2 mRNA levels and ACE2 protein levels still requires significant investigation as the interaction between transcriptional regulation and post-translational modifications of ACE2 has not been well described. Nonetheless, statin’s role in COVID-19 infection could be of interest due to its epigenetic modulation of ACE2 expression in certain organs.
Concerns for increased ACE2
It may be argued though that increased expression of ACE2 could increase the virulence of SARS-CoV-2 as there would potentially be more binding sites for the virus.3 6 In a small study of 76 patients with COVID-19, those with more severe COVID-19 symptoms had significantly higher viral loads compared with that seen from mild cases.101 Mild cases also had earlier viral clearance compared with more severe cases. It should be noted though that the severe group included significantly older patients (55.6 years vs 43.6 years). Furthermore, classification of severe was defined as respiratory distress, oxygen desaturation, partial pressure of arterial oxygen, or severe respiratory complications such as requiring mechanical ventilation, septic shock or non-respiratory organ failure. There were no deaths in either group, and no differences in the prevalence of presenting symptoms such as dyspnoea, fever or cough between groups. The only reported difference between the severe group and the mild group, besides viral load, was admission to ICU, which was 76.7% in the severe group vs 0% in the non-severe group. Therefore, it is difficult to truly ascertain from this study whether viral load was associated with clinical severity or there was selection bias because the severe group consisted of older patients who are more likely to be admitted to the ICU due to anticipation of increased viral complications. Pujadas et al102 did demonstrate that SARS-CoV-2 viral load was a statistically significant predictor of mortality, but the increased risk was very low after adjusting for clinical characteristics (only 7% increase per log-transformed copy per millilitre). Utilisation of the antiviral remdesivir did reduce the median time to recovery for COVID-19, but not mortality.103 Therefore it can be interpreted that SARS-CoV-2 viral load may have low significance in terms of mortality and COVID-19 severity.
Regardless, SARS-CoV-2 appears to be less cytotoxic when compared with SARS-CoV, as described previously. Furthermore, the characteristics associated with lower ACE2 expression such as older age, male sex and hypertension are more strongly associated with risk factors for COVID-19 severity. Conversely, the characteristics associated with higher ACE2 expression such as younger age, female sex, normotensive and possibly even Asians who smoke are all not strongly associated with COVID-19. Additionally, patients who are immunosuppressed are not necessarily at higher risk for COVID-19 infection.104 105 Therefore, we argue that we should be more concerned of the risk of COVID-19 decreasing ACE2 leading to acute lung injury, rather than COVID-19 in and of itself as a cytotoxic virus. Since SARS-CoV-2 has a relatively strong affinity to ACE2 and since SARS-CoV-2 has been shown to already be a highly contagious virus as is, increasing or decreasing ACE2 expression will most likely not affect the rates of initial infection nor viral clearance significantly.
In a small retrospective study, it was shown that ACE2 expression promoters such as ACEi or ARBs did not significantly affect the morbidity and mortality of COVID-19 infection.64 However, this study did not separate patients who used ACEi or ARBs. In a large matched case cohort study from Italy, ACEi and ARB use was more frequent among patients with COVID-19 in comparison with healthy controls, but there was no association between ACEi or ARB and COVID-19 risk. This is most likely due to the higher cardiovascular disease burden among ACEi/ARB users.106 Similarly, other antihypertensives with less ACE2 modulating capabilities (ie, calcium channel blockers, beta-blockers and diuretics) also failed to be associated with COVID-19 risk. Even though this study did not demonstrate ACEi/ARB benefits against COVID-19, ARBs did show a modest but not statistically significant association with reduced COVID-19 risk for severe or fatal cases (OR 0.83; CI 0.87 to 1.07, p>0.05). Importantly, ARB dosage was not taken into consideration and a randomised control study of different ARB dosages in patients with COVID-19 with similar cardiovascular disease profiles would be needed to adequately characterise any potential benefits of such therapy.
ARBs have more theoretical evidence for increasing ACE2 and attenuating viral-induced lung injury, and assessing only ARB’s role in COVID-19 may show benefits. Additionally, activation of ACE2 expression leads to decreased release of TGF-β and attenuation of pulmonary fibrosis, which may be a future complication following recovery from severe COVID-19.107 Therefore, increasing ACE2 expression would most likely help prevent secondary fibrotic changes following COVID-19 pneumonia. Consequently, therefore, increasing ACE2 has more theoretical benefits for patients with SARS-CoV-2 infection.
Conclusions
Based on the mechanistic understanding, certain readily available drugs targeting ACE2 expression could be repurposed to help treat COVID-19. The scientific basis behind this assumption is not derived from rigorous clinical trial evidence. However, given the gravity of the current outbreak, these theoretical drugs may find useful experimental applications.
Acknowledgments
We would like to thank all the front-line workers who have contributed greatly during this ongoing pandemic.
References
Footnotes
Contributors FC is the first author and compiled this review. SL, XX, BS, Y-YZ, AC, JN and PL provided equal contributions to different aspects of the review.
Funding This work was supported by grants from the Agencia Nacional de Investigacion y Desarrollo (ANID), Chile: FONDAP 15130011 and FONDECYT 1200490 (to SL).
Competing interests None declared.
Patient consent for publication Not required.
Ethics approval The paper is a review and did not require ethical approval from any institution.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Data sharing not applicable as no datasets generated and/or analysed for this study.