Article Text

Abstract
Background The ADP-scavenging enzyme creatine kinase (CK) is reported to reduce ADP-dependent platelet activation. Therefore, we studied whether highly elevated CK after ST-elevation myocardial infarction (STEMI) is associated with bleeding.
Methods Data of the Thrombolysis in Myocardial Infarction Study Group phase II trial on the efficacy of angioplasty, following intravenous recombinant tissue-type plasminogen activator (rt-PA), are used to assess whether peak plasma CK (CKmax) is independently associated with adjudicated fatal or non-fatal bleeding (primary) and combined bleeding/all-cause mortality (secondary) in multivariable binomial logistic regression analysis, adjusting for baseline and treatment allocation covariates.
Results The included patients (n=3339, 82% men, 88% white, mean age 57 years, SE 0.2) had a history of angina pectoris (55%), hypertension (38%) and/or diabetes mellitus (13%). CKmax ranged from 16 to 55 890 IU/L (mean 2389 IU/L, SE 41), reached within 8 hours in 51% of the patients (93% within 24 hours). Adjudicated fatal/non-fatal bleeding occurred in 30% of the patients (respectively 26% in the low vs 34% in the high CK tertile), and bleeding/all-cause mortality in 35% (29% in the low vs 40% in the high CK tertile). In multivariable regression analysis, the adjusted OR for fatal/non-fatal bleeding (vs not bleeding and survival) was 2.6 (95% CI 1.8 to 3.7)/log CKmax increase, and 3.1 (2.2 to 4.4) for bleeding/all-cause mortality.
Conclusion Highly elevated plasma CK after myocardial infarction might be an independent predictor of bleeding and haemorrhagic death. This biologically plausible association warrants further prospective study of the potential role of extracellular CK in ADP-dependent platelet activation and bleeding.
- platelets
- coronary artery disease
- emergency medicine
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Key questions
What is already known about this subject?
Evidence indicates that creatine kinase (CK), the enzyme that strongly binds ADP and converts it into ATP, reduces ADP-dependent platelet activation.
What does this study add?
Highly elevated plasma CK activity during thrombolytic and antithrombotic therapy for ST-elevation myocardial infarction is associated with a 2.6-fold higher odds of bleeding and death per log CK increase.
How might this impact on clinical practice?
Bleeding events are associated with increased mortality in acute coronary syndromes. CK might be a main risk factor for bleeding. Prospective studies will be needed to address the clinical usefulness of CK in predicting risk of non-fatal and fatal bleeding during treatment for acute coronary syndromes.
Introduction
Bleeding contributes significantly to morbidity and mortality associated with thrombolytic and antithrombotic drug therapy for ischaemic events.1–5 With the incidence of major bleeding in acute coronary syndromes (ACSs) estimated around 4%, it is well recognised that thrombolytic and antithrombotic therapy involves a fundamental tradeoff between decreasing ischaemic risk and increasing bleeding risk. Clinical assessment of bleeding risk is therefore of vital importance.1–5
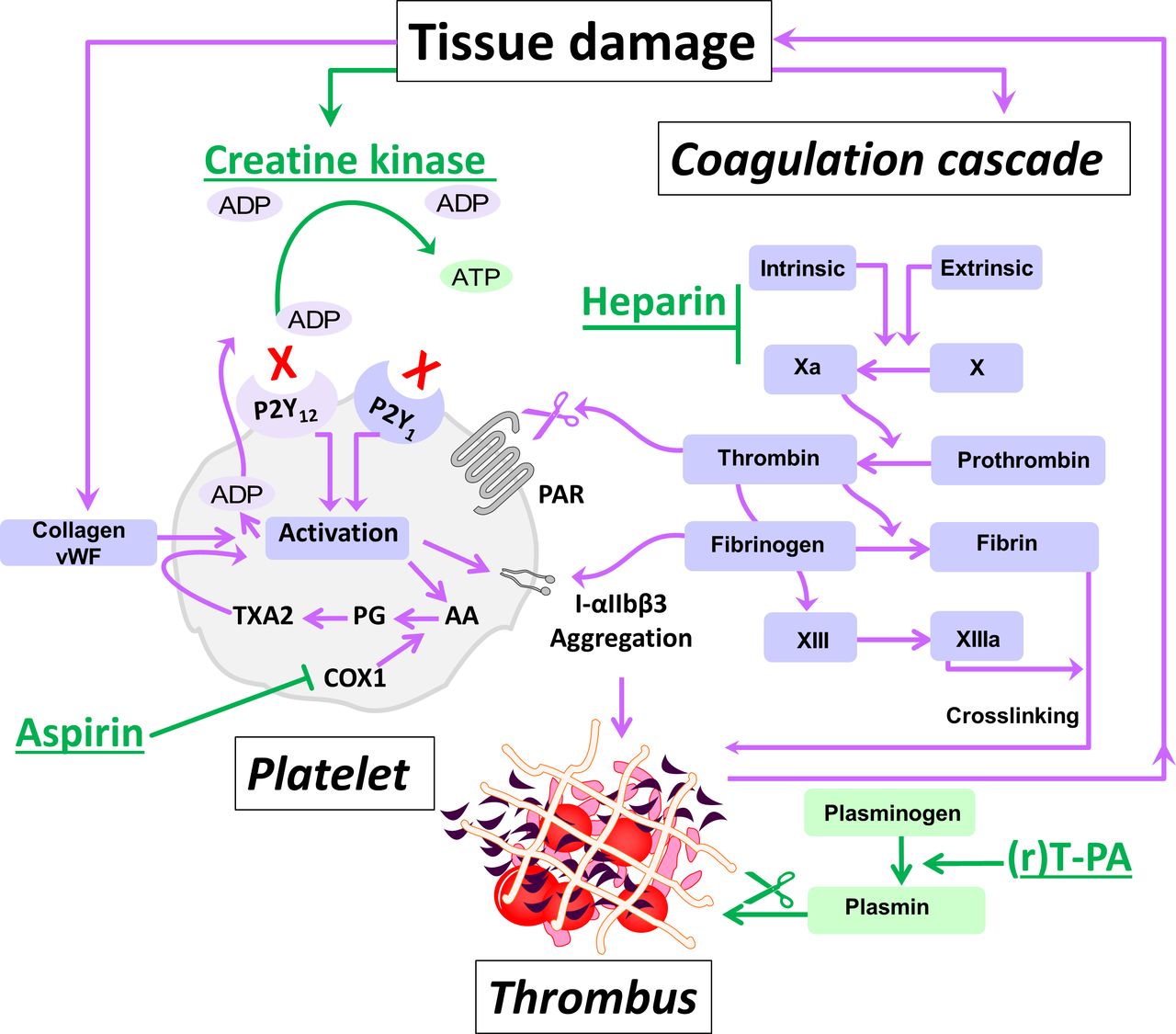
Highly elevated creatine kinase (CK) after myocardial infarction (MI) might reduce ADP-dependent platelet activation in vivo (figure 1).6–10 During myocardial ischaemia and infarction, CK and other cell constituents such as myoglobin and troponin enter plasma along with smaller molecules including phosphocreatine.1–3 6 Evidence indicates that this highly elevated CK reduces plasma ADP through its scavenging action on ADP, or through conversion into ATP, catalysing the reaction:

Highly schematic representation of thrombus formation and the proposed inhibitory action of the ADP-scavenging enzyme CK herein.6 The intrinsic and extrinsic pathways leading to thrombus formation are elaborately described elsewhere.7–10 After spontaneous plaque rupture during acute coronary syndromes and percutaneous coronary intervention, platelets adhere to the injured vessel wall, undergo shape change, cytosolic Ca2+ mobilisation and activation (through collagen and vWF). Platelet activation leads to release of ADP and TXA2 synthesised from AA through PG catalysed by COX1 (inhibited by aspirin). These secondary agonists amplify the response to injury and produce sustained platelet aggregation.7 In addition, thrombin generated by tissue damage activates platelets’ PAR. Platelet activation leads to the generation of more thrombin on the platelets’ surface, which further activates platelets, converts fibrinogen to fibrin and activates coagulation factors including factor XIII to further stabilise the platelet–fibrin clot. Plasminogen and (r)T-PA bind to the surface of the clot and plasmin degrades fibrin.7–10 ADP is considered to be central to platelet activation. ADP-stimulation of the P2Y1 receptor activates phospholipase C resulting in weak, transient platelet aggregation. Activation of P2Y12 receptor results in the activation of glycoprotein receptors IIb/IIIa (integrin (I)-αIIbβ3) and firm platelet aggregation.9 CK has a high binding capacity for ADP, which can be converted to ATP. Highly elevated extracellular CK released during major tissue damage might function to reduce ADP and dampen platelet activation, but might also lead to increased bleeding risk,6 in particular when multiple antithrombotic drugs (rt-PA, heparin, and aspirin in this study) are used. AA, arachidonic acid; COX1, cyclooxygenase 1; CK, creatine kinase; PARs, protease activated receptors; PG, prostaglandin; (r)T-PA, (recombinant) tissue-type plasminogen activator; TXA2, thromboxane A2; vWF, von Willebrand factor.
ADP +phosphocreatine <=> ATP + Creatine6
Thus, high plasma CK after MI might increase bleeding risk, in particular when drugs that affect haemostasis are used. However, once key to the diagnosis of MI, the use of troponin has led to a steady decline in CK estimation after MI and other ACS, and recent data on plasma CK in patients with MI are not readily available.1–4 11 Therefore, data of the Thrombolysis in Myocardial Infarction phase II (TIMI 2)12–17 trial were analysed to assess the association of CK with bleeding events.
Methods
Data source
TIMI 2 was a multicentre trial conducted in 24 clinical centres across the USA. The design and results of this trial, which had been completed in 1990, have been described previously.12–17 In brief, the study included 3534 patients, all treated with intravenous recombinant tissue plasminogen activator (rt-PA; Genentech, Inc, South San Francisco, California, USA) within 4 hours of the onset of chest pain, thought to be caused by ST elevation myocardial infarction (STEMI). Included patients were randomised to either an invasive strategy of cardiac catheterisation, and when anatomically appropriate, percutaneous transluminal coronary angioplasty (PTCA) or coronary artery bypass grafting (CABG); versus a conservative strategy, where these procedures were only performed in response to spontaneous recurrent or exercise-induced ischaemia.12–17 The TIMI 2 trial consisted of two major substudies combined into a main study (figure 2). The TIMI 2A study was designed to compare immediate and delayed invasive strategies, and TIMI 2B to compare immediate versus delayed beta-blocker therapy. The main study (n=3339) compared the invasive (n=1681) versus the conservative strategy (n=1658).12–17 The primary outcome was non-fatal MI (as defined by CK increase and/or ECG abnormalities) or death within 6 weeks after study entry. Major inclusion criteria were age <76 years, symptoms of ischaemic chest pain of >30 min duration and treatment possible within 4 hours of the onset of symptoms. Exclusion criteria included past or present bleeding disorder, significant intestinal bleeding, any recording of blood pressure exceeding 180 mm Hg systolic or 110 mm Hg diastolic during the presenting illness prior to randomisation, any history of cerebrovascular disease or transient ischaemic attack, oral anticoagulation therapy, advanced illness including malignancies, hepatic or renal disorders, or use of thrombolytic therapy for MI in 2 weeks before admission. During the trial, the initial dose of rt-PA of 150 mg was lowered to 100 mg after the first 520 patients, because of a relatively high number of intracerebral haemorrhages (2.1%), as judged by the Hemorrhagic Event Review Committee (HERC).18 Also, the exclusion criteria, such as history of hypertension and history of neurologic disease, were made more stringent.18 Adjunctive therapy included intravenous heparin and aspirin in all patients. Heparin was given as a 5000-unit United States Pharmacopeia bolus within 1 hour of the start of the rt-PA infusion, followed by an infusion of 1000 units per hour during 4 days guided by the activated partial thromboplastin time. Heparin was given subcutaneously, 10 000 units every 12 hours, from day 5 until hospital discharge. Aspirin dose was initially 80 mg/day starting on the same day as thrombolytic therapy, but in response to the unexpected number of intracranial haemorrhages aspirin therapy was started the day after initiation of thrombolytic therapy. The aspirin dose was increased to 325 mg/day on day 6.
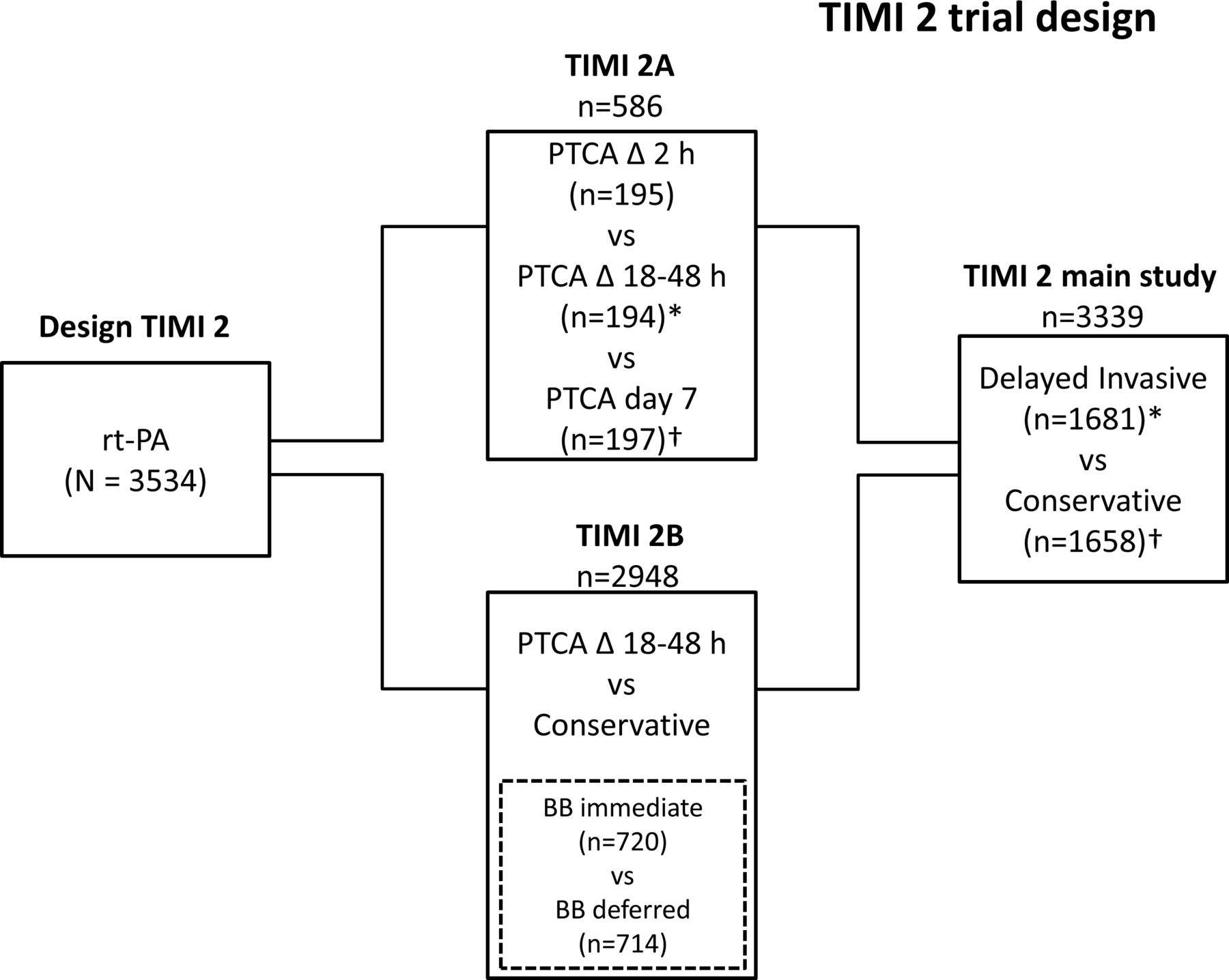
Diagram of the thrombolysis in MI phase II (TIMI 2) studies. TIMI 2 patients were treated with intravenous recombinant tissue plasminogen activator (rt-PA) within 4 hours of the onset of chest pain thought to be caused by MI, before randomisation to an invasive strategy (cardiac catheterisation, and when anatomically appropriate, PTCA or CABG within 24 to 48 hours after infarction) or conservative strategy (with invasive procedures only in response to the occurrence of spontaneous or provoked ischaemia). The trial included 3534 patients in three subsets: (1) TIMI 2 main study (large square, n=3339) in which patients were randomised between a delayed invasive strategy (n=1681) and a conservative strategy groups (n=1658); (2) TIMI 2A substudy n=586), overlapping with the main study but also including an immediate invasive strategy treatment group; and (3) the beta-blocker substudy (n=1434), a component of main study in which eligible patients were further randomised between immediate and delayed 3-blocker therapy;14 *excluding n=195 immediate invasive treatment; †including participants with PTCA on day 7 (n=197). CABG, coronary artery bypass grafting; MI, myocardial infarction; PTCA, percutaneous transluminal coronary angioplasty; TIMI 2, Thrombolysis in Myocardial Infarction phase II.12–17
Classification of haemorrhagic events and mortality
All bleeding incidents were registered by a local investigator. Furthermore, each patient with blood loss or a reduction in haemoglobin of more than 30 g/L (around 1.9 mmol/L) during hospitalisation was reviewed by the HERC. The time of onset and the bleeding site, if known, were registered. Bleeding was classified by the HERC as ‘major’ when a decrease in haemoglobin of more than 50 g/L (around 3.1 mmol/L), intracranial bleeding or cardiac tamponade was present. Bleeding was classified as ‘minor’ if a haemoglobin reduction >30 and ≤50 g/L from an identified bleeding site was present, or if the patient had spontaneous macroscopic haematuria, haemoptysis or haematemesis. Finally, patients were classified as ‘loss, no site’ if the bleeding site was unknown, but haemoglobin reduction was >40 g/L (around 2.5 mmol/L) and ≤50 g/L. Transfusion of 1 unit of packed cells or whole blood was counted as 10 g/L when assessing haemoglobin reductions. Bleeding events in trial patients who underwent surgery (mainly CABG) were considered separately.12–17 The Mortality and Morbidity Classification Committee classified all causes of death. Patients classified as having died from haemorrhage were included under ‘major haemorrhagic events.’17
Laboratory studies
Fibrinogen levels, fibrin(ogen) degradation products, rt-PA levels and plasminogen were collected from as many patients as possible at baseline, and at 50, 300 and 480 min after rt-PA infusion, and determined in a central Coagulation Core Laboratory, using standardised methods as described previously.12–17 Platelet count was determined locally at the participating centres. Total CK was estimated locally at the study centres, routinely over the first 10 days, at baseline and 4-hour intervals during the first day, at 6-hour intervals during the second day and daily at day 3 through 10. The upper reference limit (URL) of each centre was recorded.
Outcomes and sample size calculations
The main outcomes of this analysis are the independent association between peak CK activity (CKmax) with adjudicated fatal or non-fatal bleeding during hospitalisation (primary), and with adjudicated combined non-fatal bleeding and all-cause mortality (ACM, secondary). The tertiary outcome was the association of CK with all investigator reported bleeding. The probability p of events at the mean value of the CK (and other variables) was conservatively estimated to be 0.25. Sample size was estimated for two levels of the OR of disease, corresponding to an increase of 1 SD from the mean value of CK, given the mean values of the remaining variables; at OR 1.2, with at least 1138 participants needed; or at OR 2.0, with at least 95 needed for this analysis with a one-tailed alpha of 5% and 1—beta of 80.19
Data analysis
Demographic characteristics were analysed by bleeding status, and the distribution of CK during hospitalisation was assessed.12–17 Parametric versus non-parametric statistical methods were used where appropriate. However, as extreme CK values are relevant in this analysis, all values were included, with the mean and SE to the mean presented, and log transformation to the base of 10 used in regression analysis.6 The highest CK activity (IU/L) during hospitalisation in an individual patient was used (CKmax), standardised to account for the differences in URL, expressed as a normalised ratio ≥1. In addition, time to CKmax, and time to, severity and location of the first adjudicated bleeding were assessed. Bleeding events were depicted by CK tertiles, and bleeding severity by CKmax, with multivariable logistic regression analysis to assess whether log CKmax was independently associated with adjudicated fatal or non-fatal severe bleeding (primary), combined (severe) bleeding and ACM (secondary), and all investigator-reported bleeding events (tertiary), compared with no bleeding. Variables were chosen based on previous clinical data,12–17 building models adjusting for sex, ancestry (white vs non-white, as defined by the study), age, body mass index (body weight in kg divided by the squared height in metres), hypertension history, treatment assignment (conservative vs invasive), rt-PA dose, anticoagulant drugs, platelet count and a measure of haemostasis (chosen with Kendall’s tau-b correlation coefficient with adjudicated fatal or non-fatal bleeding), using forced entry and bootstrapping for internal validation. Predictive accuracy of the models was assessed with the C-statistic. Associations were also modelled excluding bleeding at puncture sites. Missing data are not imputed in the primary analysis, but depending on the number of missing data (>5%), the mechanism (ad random or not) and the data distribution, data will be considered for imputation, with the imputation strategy depending on the missing data analysis. P values are not reported in this study.20 Instead, formal statistical testing is limited and outcomes are described and depicted for inspection. Furthermore, the statistical uncertainty surrounding the estimates is communicated, where relevant through 95% CIs given between square brackets.20 Uncertainty regarding the external validity is discussed. Data in parentheses are SE unless indicated otherwise. Statistical analyses were performed with the SPSS statistical software package for Windows V.25.0 (SPSS Inc, Chicago, Illinois, USA).
Results
Baseline characteristics of the participants are depicted in table 1. Patients were predominantly white (88.3%), and men (82.1%), with a mean age of 56.8 years (SE 0.2). Patients had been randomised to the invasive strategy (n=1681) or the conservative strategy (n=1658). Median duration of hospitalisation was 10 days, with an IQR of 8 to 12 days. During hospitalisation, all 3339 patients received rt-PA (mean dose 107 mg, SE 0.3; median 100; IQR, 100 to 100), and 98.7% were given heparin, median intravenous dose for the first 5 days 21 323 U (IQR 17 383 to 25 384); and 19 540 subcutaneous for days 6 to 10 (16 820 to 21 256). Furthermore, 3201 (95.9%) received antiplatelet agents (99.4% aspirin), 77.5% as monotherapy (99.2% aspirin), 21.3% dual antiplatelet therapy (95.8% aspirin-dypiridamole) and 1.2% triple therapy. Median aspirin dose (n=3181) was 80 mg/day during the first 5 days (IQR 80 to 141 mg) and 325 from days 6 to 10 (275 to 325 mg).
Patients’ characteristics
The main findings of the TIMI trial, including recurrent infarction, cardiac function and survival have been reported previously.12–18 PTCA after rt-PA was attempted in respectively 54.0% versus 13.3% of the patients assigned to the invasive strategy versus the conservative strategy. There was no significant difference between treatment arms in the trial’s main outcome of non-fatal MI or death within 6 weeks after study entry, which occurred in 10.9% of the invasive strategy group versus 9.7% in the conservative strategy group. In addition, there was no difference in mortality between the immediate versus the deferred beta-blocker therapy groups.
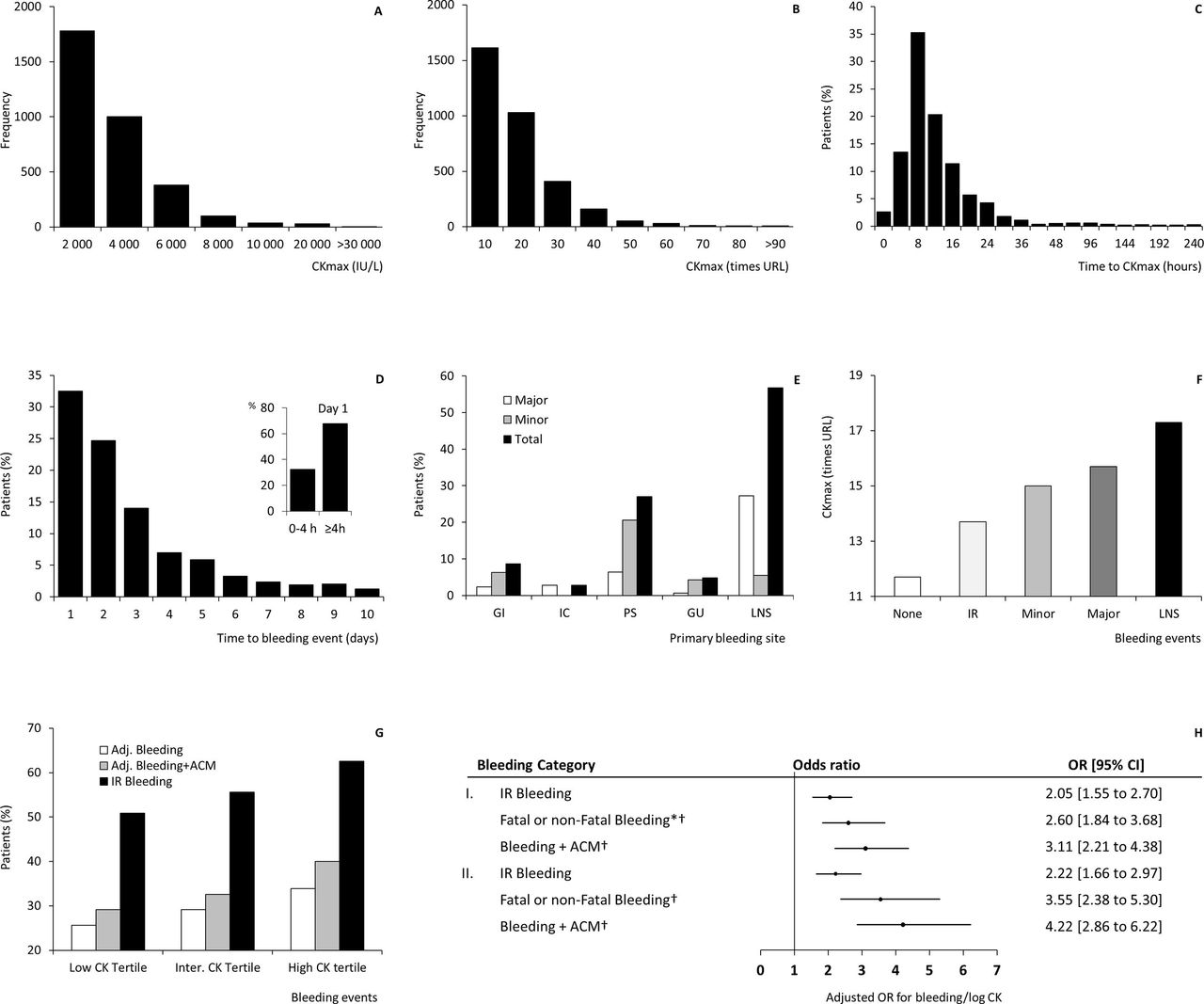
CK was estimated at 19 different time points, in total 46 523 times in 3327 patients during the hospital stay, from admission (T=0 hour) to day 10 (T=240 hour). Plasma CK ranged from <10 to 55 890 IU/L. Mean CK (SE) per patient was 900 IU/L (SE 14). CKmax ranged between 16 and 55 890 IU/L (mean 2389, SE 41), a mean of 13.5 times the URL (SE 0.2, figure 3, Panel A and B). Most patients (51.5%) had a peak in CK activity within 8 hours (figure 3, Panel C).

{kind=link}
{kind=link}
{kind=link}
Panel A to H concern the association of peak plasma creatine kinase (CKmax) with bleeding. Panel A (CK, IU/L) and B (CK normalized for the URL), show highly elevated CK activity after myocardial infarction, to more than 90 times the URL, which is thought to inhibit ADP-dependent platelet activation. Panel C and D depict the time to peak CK value (at 8 hours) and time to the first adjudicated bleeding (peak at Day 1). Panel E considers the distribution of primary adjudicated bleeding sites by severity. Panel F (n, respectively, no bleeding=1389; IR=965; Minor=361; Major=387; LNS=237) and G indicate bleeding severity by CK tertile (n=1109 per tertile, with 12 missing CK). Panel H shows the adjusted OR with (95% CI) for bleeding vs non- bleeding per log CKmax increase. I. All bleeding; II. Excluding puncture bleeding. *Primary outcome; †adjudicated bleeding events. Complete cases without imputation (n = 911 to 1428). Missing cases analysis for the multivariable binary logistic regression analyses, handling of missing data, and outcomes for other variables are reported in the online supplementary data. Adj. bleeding. First adjudicated bleeding event (fatal or non-fatalbleeding); ACM, adjudicated all-cause mortality. CI, confidence interval; CK, creatine kinase; GI, gastrointestinal; GU, genitourinary; IC, intracranial; Inter., intermediate; IR, investigator reported; LNS, loss-no-site/other; OR, odds ratio PS, puncture sites, URL, upper reference limit.
Supplemental material
Any bleeding was reported in 1950 (58.4%) patients. Of these, bleeding localisations were available in 1878, of whom 705 (37.5%) were registered to have puncture bleedings; 443 (23.6%) haematuria, and 181 (9.6%) gastrointestinal bleedings, as main primary bleeding locations. Non-surgical bleeding events as adjudicated by the HERC (first bleeding site) occurred in 985 (29.5%) of the patients (table 1), 39.3% major bleeding, 36.6% minor bleeding and 24.1% loss-no-site. Data on the time of bleeding were available in 921 (93.5%) of these patients. Adjudicated bleeding occurred on the first 2 days in 57% of the patients (figure 3, Panel D). There was a relatively high occurrence of adjudicated bleeding at vascular sites of catheter placement, venipuncture, arterial puncture or other vascular instrumentation (n=266; 27.0%; figure 3, Panel E). Blood transfusion was given to 357 (36.2%) patients with adjudicated bleeding events. Eighteen (0.5%) patients died because of bleeding, with a total ACM of 293 (8.8%) patients. It was previously reported that bleeding was associated with different factors including rt-PA dose, haemostasis variables, invasive procedures, age, female sex, low body weight and a history of hypertension.12–18 The timing of beta-blocker therapy had no apparent effect on bleeding risk.12–18 The association of bleeding with CK had not been studied previously.
CK by clinical bleeding categories is depicted in figure 3, Panels F and G. The proportion of patients with bleeding events in the low versus the high CK tertile was respectively 26% vs 34% for adjudicated fatal or non-fatal bleeding, 29% vs 40% for combined adjudicated bleeding and ACM, and 51% vs 63% for all investigator reported bleeding (figure 3, Panel G). Finally, multivariable binary logistic regression analysis suggested an independent association between CK and bleeding outcomes (figure 3, Panel H), with a twofold to fourfold increase in odds for bleeding (and ACM) compared with non-bleeding (and survival) per log CKmax increase, when holding the other variables constant. Heparin and aspirin dose, actively managed in patients with bleeding, did not significantly contribute to the model, with OR≈1. The C-index of the regression models was 0.69 [0.66 to 0.73] for the primary outcome (with 0.74 [0.70 to 0.78] for major bleeding); and 0.70 [0.67 to 0.74] for the secondary outcome. Building of the logistic regression models, model fit, handling missing data (including of fibrin(ogen) degradation product at 8 hours, having the highest correlation with bleeding of the haemostasis variables), and ORs of other variables included in the model are shown in the online supplementary data.
Discussion
The presented data indicate, to our knowledge for the first time, that plasma CK after MI is independently associated with a greater than 100% increase in the odds of fatal or non-fatal bleeding per log CKmax increase (compared with non-bleeding), when conjunctive therapy with antithrombotic and thrombolytic drugs is given. With ample experimental evidence present,6 the biological plausibility of the association is lined out in figure 1, suggesting that a massive leak of CK towards the extracellular space and circulation reduces ADP-dependent platelet activation through its binding capacity for ADP, or through conversion into ATP.6–10 The data further suggest a temporal and a dose–effect relationship, depicted in figure 3. The association is potentially clinically relevant.
Inferences from the presented data are limited. First, an association does not prove causation. Second, there is a lack of diversity in the patients, who were mainly older, white, men, with one specific condition, MI. Patients with severe hypertension, a condition known to be associated with high CK as well as with cerebral bleeding, were excluded from this trial. Thus, the strength of the association might be overestimated or underestimated in this analysis. Also, the data need to be considered with caution, as bleeding was typically multifactorial. Patients with higher CK levels probably had larger infarct sizes, and were more likely to be in a poor clinical condition, which might have affected their bleeding risk and survival. Finally, the TIMI 2 data were collected between 1986 and 1990,12–17 and it is unclear how the results might translate into contemporary clinical practice. The association reported here between CK and bleeding was not a primary objective of the TIMI 2 study, and the presented results should be considered to be hypothesis generating. The potential role of CK in haemostasis after tissue injury should be further studied and substantiated.
Tissue and endothelial factors in thrombosis and haemostasis have been extensively reviewed elsewhere (figure 1).7–10 With vascular injury, thrombus formation involves damage to the vascular wall and exposure to subendothelial molecules, initiating the coagulation cascade, adhesion of platelets, thrombus formation and thrombolysis. CK is proposed here to serve as a ‘tissue factor’ that inhibits platelet activation. Importantly, with damage to high CK tissue such as muscle and brain, large quantities of intracellular CK may be released into the extracellular space and circulation, along with ADP, ATP and phosphocreatine.6 21–24 ADP conversion or scavenging action by CK will potentially reduce ADP-dependent platelet activation,6 which might serve to reduce excessive thrombus formation. The presented data accord with a possible association between extracellular CK and inhibition of ADP-dependent platelet activation, which may act in synergy with thrombolytic or antithrombotic drugs to increase bleeding risk.
Plasma CK circulates, and although the enzyme is thought to be cleared by the liver,6 there is no known inhibitor. As a consequence, its ADP-binding effect is probably systemic. The conversion to ATP would need phosphocreatine, which is more likely to be present locally at the site of tissue damage. The role of ATP proper in platelet activation is somewhat enigmatic. ATP is reported to have either no direct effect,21 only through inhibition of ADP-dependent platelet activation,21 or act on the platelet P2X1 receptor as an inhibitor at low, and an activator at high concentrations.22 Importantly, vascular endothelium and blood cells are known to contribute to the interconversion of extracellular adenine nucleotides via ecto-nucleoside triphosphate diphosphohydrolases, ecto-nucleotide pyrophosphatase/phosphodiesterases, ecto-5'-nucleotidase and alkaline phosphatases.23 24 Thus, the effect of highly elevated extracellular and circulating CK on platelet activation is thought to be mainly through ADP reduction, but the role of ATP, and the potential interaction between CK and ectoenzymes that convert ATP and ADP needs further exploration.
Although the association between CK and platelet function was not assessed in this study, existing evidence indicates that CK inhibits platelet aggregation,6 and distinct hypoaggregability of platelets or increased bleeding risk were reported immediately after MI.17 25 Furthermore, the ill-understood ‘trauma-induced coagulopathy’ may ensue after major trauma of CK-rich tissue (muscle or brain), associated with inhibition of ADP-dependent platelet aggregation and intractable bleeding.26 Also, bleeding events occur more frequently in population subgroups with relatively high tissue and plasma CK, such as in persons of (West-)African ancestry.6 27–32 When given fibrinolytic therapy for STEMI, this group had a higher risk of moderate or severe bleeding, adjusted OR 1.36 [1.14 to 1.62], with a HR of 2.83 [2.08 to 3.86] for haemorrhagic death,30 and a higher bleeding risk was observed with the use of clopidogrel (adjusted HR 3.78 [1.35 to 10.60]),31 as well as with dabigatran.32
Bleeding is a very relevant clinical issue in thrombolytic and antithrombotic therapy for acute ischaemic syndromes, and a major cause of morbidity and mortality.1–5 With intensification of antiplatelet therapy, adding a P2Y12 inhibitor to aspirin monotherapy as well as prolongation of dual antiplatelet therapy to prevent ischaemia,2 it has become of vital importance to identify patients at high risk for bleeding. Plasma CK might aid in this risk stratification. The observed association between CK and bleeding is proposed to be present regardless of the definition of ACS or the choice of treatment. Since TIMI 2 was conducted, the use of troponin (cTnI and cTnT) altered the definition of MI.33 Although currently not routinely assessed, plasma CK activity may be highly elevated in ACS, including STEMI and non-STEMI, or after percutaneous coronary intervention (PCI) periprocedural myocardial injury or infarction.33–35 While all patients in TIMI 2 received rt-PA, primary PCI is now preferred,3 but the association could be suspected in any patient with high CK, whether because of tissue trauma,26 (West-)African ancestry,6 or use of drugs that increase CK (such as statins), and in particular with the concurrent use of drugs that prevent thrombosis or lyse the clot.17 30–32 CK may be the elephant in the room in these conditions, apparently overlooked as a major ADP scavenger molecule in the extracellular space and the circulation. High plasma CK could be relevant in every clinical context of bleeding.6
However, more data are needed to substantiate this potential role of circulating CK. Does CK proper inhibit platelet activation in vivo, and how high should CK be to increase bleeding risk? Is such effect local or systemic, how is CK inhibited, and what is the effect of ATP, if any? Is there an interaction with antithrombotic and thrombolytic medication, is plasma CK clinically useful to stratify bleeding risk and will dose reduction of antithrombotic and thrombolytic drugs reduce the bleeding events and death associated with high CK? These important questions need answers. It is hoped that presenting the theory and these associative data will revive the estimation of CK in patients with ACS to study the association with bleeding, in particular when antithrombotic or thrombolytic drugs are used.
Acknowledgments
The manuscript was prepared using TIMI 2 research materials obtained from the NHLBI Biologic Specimen and Data Repository Information Coordinating Center. The content does not necessarily reflect the opinions or views of the TIMI 2 study members or the NHLBI.
References
Footnotes
Contributors LMB helped in conceptualisation, data curation, formal analysis, investigation, methodology, project administration, resources, supervision, validation, visualisation, writing original draft, review and editing. JF helped in conceptualisation, investigation, methodology, resources, validation, visualisation, review and editing.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests LMB is an inventor on patent WO/2012/138226 (filed).
Patient consent for publication Not required.
Ethics approval All patients had given written informed consent to be included in the study. The current analysis was approved by the review board of the Research Ethics Committee of the Radboud University Nijmegen Medical Centre on 30 April 2019 (registration number 2019–5356), and by the US National Heart, Lung, and Blood Institute (RMDA V02 1d20120806) on 6 May 2019.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Data are available in a public, open access repository. Data are available at the Biologic Specimen and Data Repository Information Coordinating Center (BioLINCC) supported by the National Heart, Lung, and Blood Institute of the United States National Institutions of Health (NIH), USA. https://biolincc.nhlbi.nih.gov/home/