Article Text

Abstract
Objective The myosin-binding protein C (MYBPC3) c.927-2A>G founder mutation accounts for >90% of sarcomeric hypertrophic cardiomyopathy (HCM) in Iceland. This cross-sectional observational study explored the penetrance and phenotypic burden among carriers of this single, prevalent founder mutation.
Methods We studied 60 probands with HCM caused by MYBPC3 c.927-2A>G and 225 first-degree relatives. All participants underwent comprehensive clinical evaluation and relatives were genotyped.
Results Genetic and clinical evaluation of relatives identified 49 genotype-positive (G+) relatives with left ventricular hypertrophy (G+/LVH+), 59 G+without LVH (G+/LVH−) and 117 genotype-negative relatives (unaffected). Compared with HCM probands, G+/LVH+ relatives were older at HCM diagnosis, had less LVH, a less prevalent diastolic dysfunction, fewer ECG abnormalities, lower serum N-terminal pro-B-type natriuretic peptide (NT-proBNP) and high-sensitivity cardiac troponin I levels, and fewer symptoms. The penetrance of HCM was influenced by age and sex; specifically, LVH was present in 39% of G+males but only 9% of G+females under age 40 years (p=0.015), versus 86% and 83%, respectively, after age 60 (p=0.89). G+/LVH− subjects had normal wall thicknesses, diastolic function and NT-proBNP levels, but subtle changes in LV geometry and more ECG abnormalities than their unaffected relatives.
Conclusions Phenotypic expression of the Icelandic MYBPC3 founder mutation varies by age, sex and proband status. Men are more likely to have LVH at a younger age, and disease manifestations were more prominent in probands than in relatives identified via family screening. G+/LVH− individuals had subtle clinical differences from unaffected relatives well into adulthood, indicating subclinical phenotypic expression of the pathogenic mutation.
- cardiomyopathy hypertrophic
- genetics
- echocardiography
This is an open access article distributed in accordance with the Creative Commons Attribution 4.0 Unported (CC BY 4.0) license, which permits others to copy, redistribute, remix, transform and build upon this work for any purpose, provided the original work is properly cited, a link to the licence is given, and indication of whether changes were made. See: https://creativecommons.org/licenses/by/4.0/.
Statistics from Altmetric.com
Key questions
What is already known about this subject?
Hypertrophic cardiomyopathy (HCM) is the most common monogenic heart disease, with diverse genetic and phenotypic expression. More than 2000 causative mutations in at least eight genes have been described in association with HCM. Although mutations in myosin-binding protein C (MYBPC3) represent 30%–40% of all HCM mutations, most of the causal mutations are usually unique to one or few families.
What does this study add?
A prevalent MYBPC3 founder mutation is responsible for >90% of sarcomere HCM in Iceland, providing valuable opportunity to explore associations between genotype and clinical phenotype. To our knowledge, this is the largest and most comprehensive genotype–phenotype study that exists on a single HCM mutation. Phenotypic manifestations of the Icelandic MYBPC3 founder mutation varied by age, sex and proband status, and were remarkably similar to other distinct founder populations with truncating MYBPC3 variants, suggesting a common effect in the context of haploinsufficiency in the myosin-binding protein.
How might this impact on clinical practice?
Our findings underscore the need for lifelong surveillance of male and female MYBPC3 mutation carriers, and identifying factors that modify disease expression and drive phenotypic progression.
Introduction
Hypertrophic cardiomyopathy (HCM) is a genetically heterogeneous disorder with highly variable clinical expression. Although most HCM mutations occur in two cardiac sarcomere protein genes, MHY7 (encoding β-myosin heavy chain) and MYBPC3 (encoding myosin-binding protein C), specific pathogenic variants are usually unique to one or a few families. The small numbers of individuals with identical HCM mutations, the great diversity in background genotypes and the substantial phenotypic heterogeneity has limited the ability to determine the impact of genotype on phenotype and clinical outcomes. Studying populations with founder mutations helps address some of these challenges, but to date, only a few analyses have systematically described HCM phenotypes caused by founder mutations in at-risk relatives.1 2
We previously reported an Icelandic MYBPC3 founder mutation, c.927-2A>G, that was introduced ~500 years ago and has an estimated population prevalence of 0.36%. This single mutation is responsible for >90% of sarcomeric HCM in Iceland,3 a geographically isolated country with a relatively homogeneous population. Here, we explored the inheritance and clinical consequences of the Icelandic MYBPC3 founder mutation in 60 HCM probands and 225 first-degree relatives. Because the prevalence of the Icelandic variant is higher than that seen in other single founder variants,1 4–7 it provides a valuable opportunity to explore associations between genotype and clinical phenotype at a larger scale.
Methods
Study design and population
This study used a cross-sectional observational cohort design. HCM probands had a previously established HCM diagnosis and were found to carry the MYBPC3 c.927-2A>G founder mutation through a nationwide genetic study of HCM in Iceland.3 deCODE Genetics genealogical database was used to identify all first-degree relatives. None of the relatives were previously diagnosed with HCM. Relatives were grouped according to genotype and the presence or absence of left ventricular hypertrophy (LVH; defined as a LV wall thickness (LVWT) ≥12 mm in adults or a z-score of ≥2 in children) as follows: mutation carrier with LVH (G+/LVH+), mutation carrier without LVH (G+/LVH−) and healthy mutation-negative (unaffected).
Participants underwent study visit between November 2012 and June 2014, including assessment of medical history and heart failure symptoms (New York Heart Association functional class), blood pressure, ECG and standardised echocardiography. Blood was drawn for genetic analysis and serum biomarker analysis. Subjects with known hypertension, cardiovascular disease other than HCM or medical conditions associated with increased collagen turnover were excluded. All investigators were blinded to the subjects’ clinical and genetic status. This study complied with the Declaration of Helsinki and was approved by the National Bioethics Committee of Iceland and the Icelandic Data Protection Authority. Informed consent was obtained from all participants, and parental consent was obtained for participants under 18 years of age.
Genetic analysis
Genomic DNA, extracted from whole-blood samples, was obtained from all relatives and genotyped to determine the presence or absence of the MYBPC3 c.927-2A>G mutation, as previously described.3
Echocardiography
Transthoracic echocardiograms were obtained using Vivid E9 ultrasound systems (GE Healthcare, Horten, Norway), including two-dimensional images, spectral, colour and tissue Doppler interrogation following a standardised protocol. The average of three cardiac cycles was used for measurements of cardiac dimensions, mitral inflow patterns and myocardial velocities according to published guidelines.8 9 Maximal LVWT was defined by the maximal LVWT in any of the LV segments measured in the long-axis or short-axis parasternal view. Relative wall thickness was defined as two times the posterior wall thickness divided by the LV end-diastolic diameter (LVEDD). Left atrial (LA) volumes were measured using the biplane disk summation at end-systole. To allow comparisons between subjects with different body sizes, chamber measurements were reported as indexed to body surface area (BSA). The peak instantaneous LV outflow tract gradient was measured at rest and during the Valsalva manoeuvre with continuous-wave Doppler. The pattern of LV septal hypertrophy was divided into the following morphological subtypes: reverse curvature, sigmoid, neutral and apical.10 Diastolic function was assessed and graded as normal or diastolic dysfunction grades 1–3 based on expert consensus recommendations.9 Images were analysed offline using Echopac BT 112 software (GE Healthcare, Horten, Norway). Echocardiograms were analysed by two observers with expertise in cardiac imaging (BA, GTG) without knowledge of genotypes.
ECG
Standard 12-lead ECGs were obtained at rest in the supine position. All ECGs were analysed by a single investigator (BA) according to standard criteria as described elsewhere.11 The Sokolow–Lyon LVH voltage criteria were defined as SV1 + RV5 or RV6 ≥35 mm. The Cornell LVH voltage criteria were defined as a RaVL+ SV3>28 mm for men and >20 mm for women. Conduction disturbance included interventricular conduction delay (QRS duration ≥110 ms), left bundle branch block or right bundle branch block. QST was defined as the presence of pathological Q waves, T wave inversion or ST-segment depression.11
Serum biomarkers
Plasma concentrations of N-terminal B-type natriuretic peptide (NT-proBNP), high-sensitivity troponin I (hsTnI) and carboxy-terminal propeptide of procollagen type I (PICP) were measured. Plasma concentrations of NT-proBNP and hsTnI were measured in the Biomarker Research and Clinical Trials Laboratory at Brigham and Women’s Hospital, Boston, MA. NT-proBNP was measured using the proBNPII immunoassay (Roche, Indianapolis, IN, USA) with a coefficient of variation (CV) of 3.8% at 127 pg/mL and 2.4% at 4180 pg/mL. hsTnI was measured using an ultrasensitive immunoassay utilising a single-molecule counting technology (Erenna hsTnI, Singulex, Palo Alto, CA, USA) with a CV of 12.5% at 6.8 ng/L and 13.7% at 39.2 ng/L. PICP was measured at the CIMA of University of Navarra, Pamplona, Spain, using the METRA Enzyme Immunoassay (Quidel Corporation) with an inter and intra-assay CV of 6.3% and 6.4%, respectively, and a lower limit of detection of 0.2 ug/L. All assays were performed using commercially available reagents by technicians blinded to clinical and genetic statuses of the subjects. Details have been described previously.12 13
Statistical analysis
Descriptive data are presented as the means±SD or counts and proportions. Baseline characteristics were compared with Student’s t test for continuous variables, and the χ2 test or Fisher’s exact test for categorical variables, as appropriate. Generalised linear models were used to examine differences in echocardiographic measures, ECGs and biomarkers, adjusting for age, sex and within-family correlation, where correlation between relatives was assumed to be proportional to their kinship. Values are expressed as the adjusted means (SE). Biomarkers were log-transformed due to skewed distributions, and values below the detectable threshold were set to the lower detection limit (0.1 pg/mL; n=9 for hsTnI). Pearson correlation coefficients were calculated to assess correlations between continuous variables. P values of <0.05 were considered statistically significant.
Results
Study population
We studied 285 individuals, including 60 HCM probands (mean age, 53±17 years) described previously3 and 225 first-degree relatives (figure 1). Of the relatives, 108 (48%) were founder mutation carriers (G+; mean age 41±20 years) and 117 (52%) were healthy mutation-negative individuals (unaffected; mean age, 40±18 years). Among the G+ relatives, 49 (45%) were G+/LVH+ (mean age, 50±19 years) and 59 were G+/LVH− (mean age, 34±17 years).
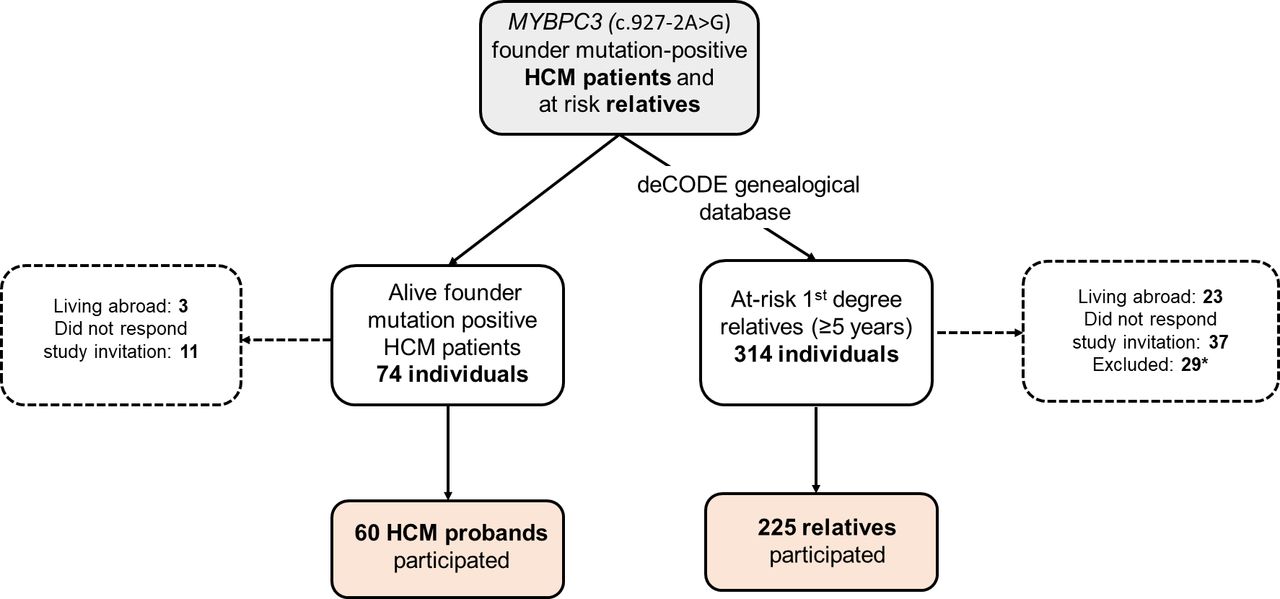
A flow diagram demonstrating the study cohort. *27 individuals were excluded due to hypertension and/or coronary artery disease, and 2 because of metastatic cancer.
Baseline clinical characteristics
Baseline demographics are summarised in table 1. HCM probands and G+/LVH+ relatives were predominantly men (67% and 55%, respectively). The probands were diagnosed with HCM at younger ages (41±14 years) than G+/LVH+ relatives (50±19 years; p value=0.004) and had more symptoms (48% HCM probands vs 4% G+/LVH+ relatives were New York Heart Association (NYHA) functional class ≥II; p<0.0001). Although the majority (96%) of G+/LVH+ relatives were asymptomatic, one (8.2%) had a history of atrial fibrillation, four (8.2%) had experienced unexplained syncope and one (2%) had been hospitalised for symptomatic heart failure.
Baseline clinical characteristics by study group
G+/LVH− relatives were predominantly women (68%); 98% had no symptoms (NYHA class I) and none had a history of atrial fibrillation.
Echocardiographic findings
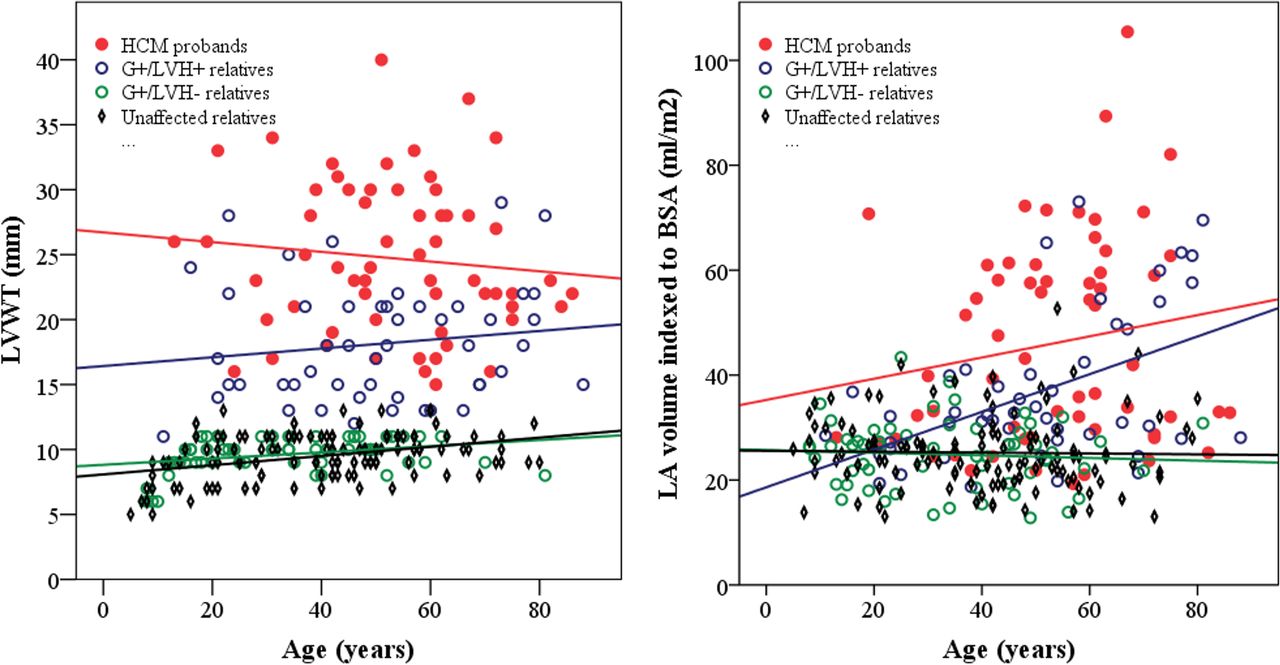
Echocardiographic findings are summarised in table 2. There was considerable variation in LVWT (figure 2A) among HCM probands and G+/LVH+ relatives across all ages, but the HCM probands had ~40% greater maximal LVWT overall. HCM probands also exhibited greater variation in LA size and larger mean LA volumes than their G+/LVH+ relatives (figure 2B). Furthermore, HCM probands had higher LV outflow tract gradients, LVWT to LVEDD ratios and more prominent diastolic abnormalities (figure 3) than G+/LVH+ relatives. Cardiac morphology showed a reverse curvature septum in 90% of the HCM probands, compared with 69% of the G+/LVH+ relatives (p=0.019), while 7% of the probands and 22% of the G+/LVH+ relatives had neutral septum morphology (p=0.040). Sigmoid septum morphology or apical hypertrophy was rare in both groups.
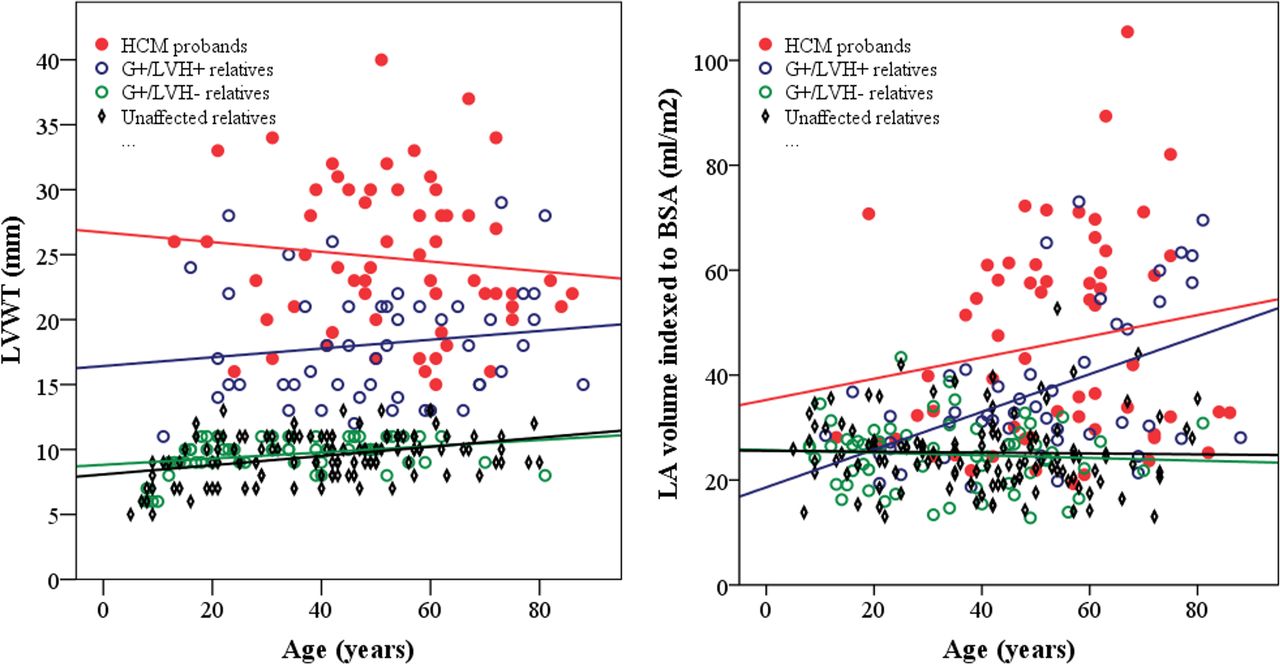
Scatter plots stratified by genotype–phenotype showing the relationship between (a) age and LVWT. There was no correlation between age and LVWT in HCM probands (red line, r=−0.11, p=0.42) and G+/LVH+ relatives (blue line, r=0.14, p=0.32). A modest linear increase in LVWT by age was noted in G+/LVH− relatives (green line, r=0.32, p=0.015) and unaffected relatives (black line, r=0.38, p<0.0001). (b) Age and LA volume indexed to BSA. There was a significant relationship between LA volume and age in G+/LVH+ relatives (blue line, r=0.49, p=0.0003), while there was no significant correlation among HCM probands (red line, r=0.17, p=0.19), G+/LVH- relatives (green line, r=−0.07, p=0.61) and unaffected relatives (black line, r=−0.02, p=0.81).

Stacked column bars demonstrating LV diastolic function for each group. HCM probands had significantly worse diastolic function than their G+/LVH+ relatives. Diastolic function was normal in 38% of HCM probands, 71% of G+/LVH+ relatives, 97% of G+/LVH− relatives and 95% of unaffected relatives. P values were adjusted for age, sex and within-family correlation.
Echocardiographic findings and biomarker analysis
The mean LVWT was slightly higher in G+/LVH− compared with unaffected relatives (9.9±0.2 vs 9.5±0.1, p=0.032), as was the relative wall thickness and ratio of LVWT to LVEDD (table 2). However, when compared across the age span of all study participants, neither LVWT nor LA volumes were substantially different between G+/LVH− and unaffected relatives (figure 1A and B). Similarly, diastolic function (figure 2) did not distinguish G+/LVH− relatives from unaffected relatives.
Serum biomarkers
NT-proBNP (reflecting haemodynamic stress) and hsTnI (reflecting myocardial injury) levels were significantly higher in HCM probands than in G+/LVH+ relatives (table 2). NT-proBNP levels were higher in G+/LVH− relatives than in unaffected relatives, but hsTnI and PICP (reflecting collagen synthesis) levels were comparable.
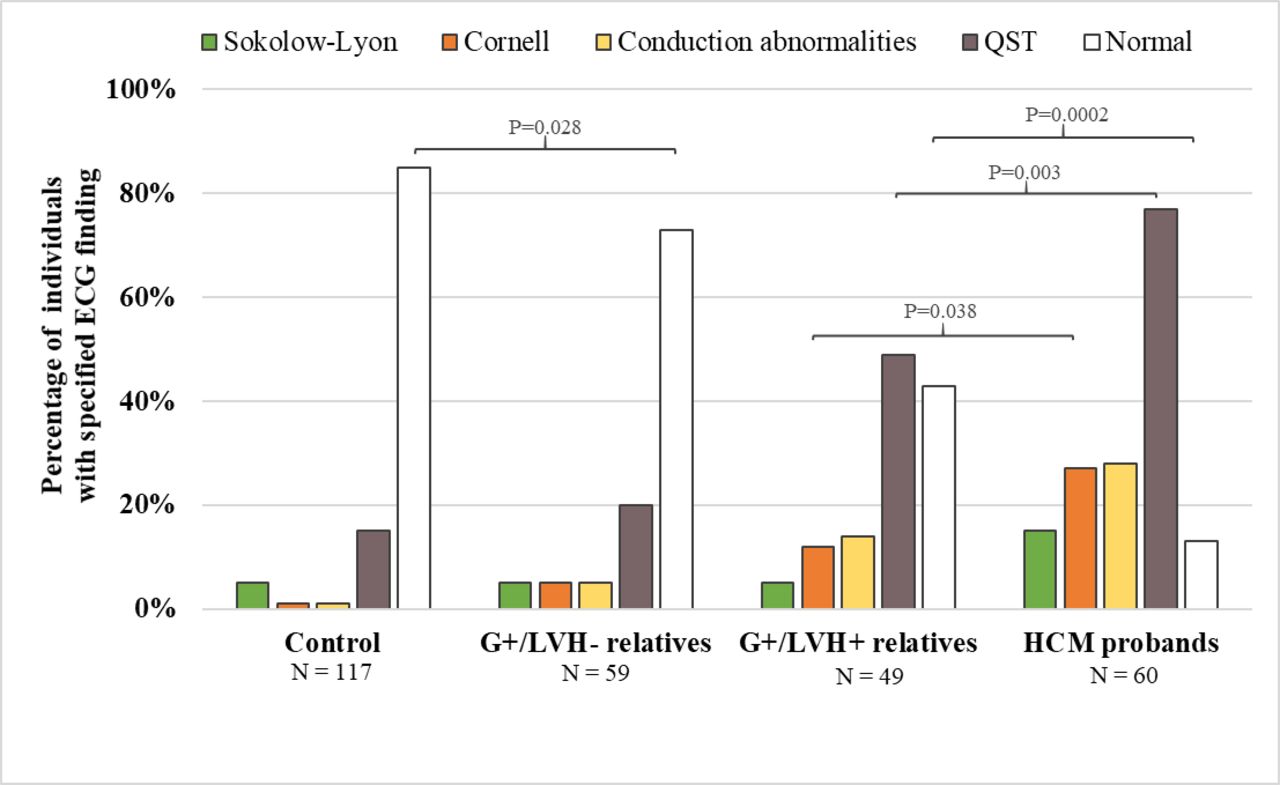
ECG
ECGs were abnormal in 87% of the HCM probands and 57% of the G+/LVH+ relatives (p=0.0002; figure 4). In contrast, 27% of the G+/LVH− relatives and 15% of the unaffected relatives had abnormal ECGs (p=0.028). Repolarisation abnormalities and/or Q waves (QST) were the most frequently observed abnormalities and were present in 77% of G+/LVH+ probands and 49% of G+/LVH+ relatives (p=0.003). QST was present in 20% of G+/LVH− relatives and 15% of unaffected relatives (p=0.07). The Sokolow–Lyon and Cornell voltage criteria had poor sensitivity for identifying LVH in HCM probands (15% and 27%, respectively) and G+/LVH+ relatives (8.2% and 12%, respectively).

Frequency of ECG abnormalities among each group. ECG abnormalities, including Cornell voltage criteria, Q waves and/or repolarisation abnormalities (QST), were significantly more prevalent among HCM probands than among their G+/LVH+relatives. The G+/LVH- relatives were less likely to have a normal ECG than their unaffected relatives. P-values were adjusted for age, sex and within-family correlation. P-values<0.05 are shown for comparison between HCM probands and G+/LVH+relatives, as well as G+/LVH- relatives vs controls.
Penetrance
The penetrance of LVH in G+family members (n=108) was age- and sex-dependent (figure 5). LVH was identified in G+/LVH+ relatives at ages spanning from 11 to 88 years, with a maximal LVWT ranging from 11 to 29 mm. Among G+ relatives aged under 40 years, 39% of men but only 9% of women had LVH (p=0.015). In contrast, after 60 years of age, the prevalence of LVH was high (83%–86%) in both sexes, but not complete. The oldest G+/LVH− male was 70 years old, and the oldest G+/LVH− female was 81 years old.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Clustered bar chart demonstrating the proportion (and 95% CI) of MYBPC3 c.927-2A>G mutation carriers identified through family screening with clinical evidence of LVH, by age and gender.
Discussion
Analyses of cardiac imaging, ECG and serum biomarker studies in a nationwide cohort provided insights into the clinical expression and penetrance of a highly prevalent Icelandic HCM founder mutation, MYBPC3 c.927-2A>G. Among the G+ relatives, 45% had LVH at the time of the study. Female mutation carriers developed LVH later in life than males. Before the age of 40 years, the penetrance of LVH was 9% in G+ females in contrast to 39% in G+ males. However, LVH was present in >80% in both sexes after the age of 60 years, indicating a high lifetime penetrance of HCM. Although HCM probands and G+/LVH+ relatives were of comparable ages at the time of the study, HCM probands had a greater burden of disease, including more severe hypertrophy and diastolic dysfunction, more arrhythmias and clinical symptoms, and higher serum troponin and natriuretic peptide levels. G+/LVH− individuals had subtle differences in LV geometry compared with their unaffected relatives, a higher prevalence of ECG abnormalities and a modest, intra-normal elevation of NT-proBNP levels. Collectively, these findings provide evidence for early LV remodelling and haemodynamic stress prior to the development of LVH.
Age- and sex-related penetrance
There was substantial variation in age of diagnosis and the degree of LVH among the G+ relatives identified through family screening that appears to be associated with both age and sex. LVH was rarely seen in children or adolescents carrying this MYBPC3 founder mutation. Our results confirm the male predominance and earlier onset of clinical disease previously described in HCM.14–16 However, we make a novel observation that this imbalance attenuates with advancing age. Notably, the prevalence of LVH in female mutation carriers increased considerably after the age of 60 years, indicating an approximate 20-year delay in the penetrance of clinically detectable disease for female G+ relatives. Older male and female mutation carriers had an equal prevalence of LVH, indicating that the Icelandic founder mutation is associated with a high lifetime penetrance of HCM. Whether these differences are related to long-term effects of endogenous oestrogens or other factors is unclear.17 Additionally, identifying factors that render certain mutation-carriers resilient to developing HCM, even at advanced ages, is a high priority.
Relatives with LVH
The Icelandic G+/LVH+ relatives diagnosed through family screening had less advanced disease (few symptoms, lower maximal LVWT, smaller LA, less obstructive physiology and less prevalent diastolic dysfunction) despite older age at diagnosis compared with HCM probands. In addition, the prototypical HCM morphology of reverse curvature septal hypertrophy was less common in G+/LVH+ relatives than in the probands (p=0.019). Assessments of ECG abnormalities, systolic and early diastolic tissue Doppler velocities, and hsTnI and NT-proBNP confirmed milder clinical expression in G+/LVH+ relatives than in HCM probands.
These findings are a vivid demonstration of the phenotypic variability possible with a single pathogenic mutation. Factors that drive the marked heterogeneity in disease expression and clinical outcomes are poorly understood. By anchoring to a single pathogenic variant, further detailed studies of founder populations will provide valuable opportunities to better refine genotype–phenotype correlations and to better understand features that contribute to phenotypic heterogeneity.
Comparisons with other studies of MYBPC3 founder mutations
Multiple founder mutations have been identified in MYBPC3,1 4–7 that all cause premature truncation and haploinsufficiency of myosin-binding protein in sarcomeres. The effects of three Dutch founder mutations (46% MYBPC3 c.2373dupG; 32% MYBPC3 c.2827C>T; 22% MYBPC3 c.2864_2865delCT) were characterised in 134 HCM probands and 54 G+/LVH+ relatives, identified by family screening,2 similar to our approach. An Italian study of the MYBPC3 p.F305Pfs*27 founder mutation1 characterised 19 HCM probands and 29 G+/LVH+ relatives who were identified either by family screening (72%) or clinical presentation (28%). Across these founder mutation populations, HCM probands had a similar predominance of male sex, age of clinical diagnosis, maximum LVWT and prevalence of NYHA ≥2 functional class. Among Dutch and Icelandic G+/LVH+ relatives diagnosed by family screening, clinical features were also remarkably consistent and milder than those in HCM probands (table 3). Although previous studies underscore the heterogeneous nature of HCM even among individuals with identical mutations18 and attribute these differences to genetic background and/or modifying environmental factors and lifestyles, we observed remarkable phenotypic similarities among cohorts of HCM probands and G+/LVH+ relatives with different MYPBC3 founder mutations. Given the distinct genetic backgrounds of the Dutch and Icelandic populations, differences in environments and lifestyle, these data provide evidence for relatively consistent direct or proximal consequences of MYBPC3 haploinsufficiency. However, the concordance between G+/LVH+ relatives may also indicate the presence of other common genetic modifiers that attenuates disease expression (relative to probands) despite the presence of pathogenic genotypes. Given the shared molecular mechanisms between different truncating MYBPC3 founder mutations and the statistical power achieved by combining multiple families with these mutations, collaborative genomic analyses may uncover important deleterious or protective alleles.
Comparison of MYBPC3 founder mutations in three populations
Preclinical HCM and early phenotypes
Early morphological and electrophysiological phenotypes reported among sarcomere mutation carriers with normal LVWT include subtle changes in LV cavity size and geometry, impaired LV relaxation19–22 and ECG abnormalities.11 As most of these studies analysed small and/or heterogeneous cohorts, interpretation of these findings has been difficult.20 23 Our analyses of preclinical HCM subjects with a shared founder MYBPC3 mutation advances these findings. In all, 59 G+ relatives had no diagnostic criteria for HCM, including 10 individuals over 50 years of age. LVWT and LA dimensions were normal, although the ratio of LVWT to LVEDD and the relative wall thickness was slightly higher in G+/LVH− relatives compared with unaffected relatives. Diastolic function parameters were normal in 97% of G+/LVH and 95% of unaffected relatives, and no significant difference was detected in any parameter of LV relaxation and estimation of LV filling pressures. However, while NT-proBNP levels did not discriminate preclinical (G+/LVH−) subjects from controls in prior studies,12 13 20 23 24 levels were higher in our G+/LVH− compared with unaffected relatives, although still within the normal range. ECG abnormalities, predominantly Q waves and repolarisation changes, were more prevalent among G+/LVH− individuals than among unaffected relatives (27% vs 15%; p=0.028) and may reflect early manifestations of HCM,11 although with low positive predictive value. Overall, the composite phenotypic manifestations of these 59 G+/LVH− adults were subtle. We suggest that analyses of the impact of genetics and lifestyle, and determination of how early phenotypic changes relate to development of clinically overt HCM are worthy of further study.
Limitations
This was a cross-sectional study, and participants were only screened for the MYBPC3 c.927-2A>G mutation. Therefore, we cannot exclude the possibility of additional variants that may contribute to clinical variability. This study involved families with at least one member with overt HCM due to the MYBPC3 c.927-2A>G founder mutation, while we estimate that~85% of the founder mutation carriers in Iceland or approximately 950 individuals remain clinically undetected with unknown clinical status.3 Similar to most HCM studies, this study is biassed towards individuals with clinical HCM diagnosis and their first-degree relatives, and it might be difficult to extrapolate the results SE findings to the whole founder population of Iceland. A larger population-based study would add to the current knowledge of disease penetrance and disease expression and possibly identify either genetic or environmental modifiers that have significant impact on disease manifestation.
Conclusions
Through comprehensive family screening of a nationwide cohort of Icelandic individuals carrying the same MYBPC3 founder mutation, we demonstrated that HCM occurs at an earlier age in men, but the lifetime penetrance appears to be high and equivalent in both men and women. Relatives diagnosed with HCM in the context of family screening were older at initial evaluation and had a milder clinical phenotype than HCM probands. Furthermore, phenotypic manifestations of the Icelandic MYBPC3 founder mutation were remarkably similar to other distinct founder populations with truncating MYBPC3 variants suggesting a common effect in the context of haploinsufficiency in the myosin-binding protein. Finally, we found a higher prevalence of abnormal ECGs and subtle differences in LV geometry in G+/LVH– subjects compared with unaffected relatives. Collaborative longitudinal clinical and genetic studies are needed to identify factors that modify disease expression, drive phenotypic progression and lead to adverse clinical outcomes.
Acknowledgments
We thank Daniel F. Gudbjartsson and Karl Ove Hufthammer for providing statistical assistance and Elizabeth Cook, Lilja Pétursdóttir and Sigurbjörg Sigurdardottir for their assistance with data collection.
References
Footnotes
CES, CYH and GTG are joint senior authors.
Contributors Conceptualisation: BA, GTG, CYH, CES, JGS and BJM. Data curation: BA and GTG. Formal analysis: BA, GTG and JGS. Funding: GTG, CYH, CES, JGS and BA. Investigation: BA, GTG, CYH, CES, JGS, MAB, BL, JD and PJ. Methodology: BA, GTG, CYH, CES and JGS. Project administration: BA, GTG, CYH, CES and JGS. Resources: GTG, CYH, CES, JGS, BA and RD. Supervision: GTG, CYH, CES and JGS. Validation: BA, GTG, CYH, CES and JGS. Visualisation: BA, GTG, CYH, CES and JGS. Writing: original draft: BA, GTG, CYH and CES. Writing: review and editing: All authors. BA and GTG are responsible for the overall content as guarantors.
Funding This study was supported by the Howard Hughes Medical Institute (CES), the National Institutes of Health (5HL084553: CES, JGS; 1P20HL101408: CYH; 1P50HL112349: CYH), Akureyri Hospital Research Fund (GTG), Landspitali–The National University Hospital of Iceland Research Fund (BA), the Icelandic Cardiac Society Research Fund (GTG).
Competing interests CES and JGS are founders and own shares in Myokardia Inc., a startup company that is developing therapeutics that target the sarcomere. CYH is a consultant for Myokardia, Inc. Myokardia had no involvement in this study.
Patient consent for publication Not required.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Data are available on reasonable request, including deidentified individual participant data.