Article Text

Abstract
Introduction Cardiomyopathies are a heterogeneous heart diseases group in terms of morphology and aetiology. Hypothesising a tropical specificity and given an absence of data in French Guiana, the primary objective of our study was to describe morphologies and aetiologies of cardiomyopathies observed at Cayenne General Hospital.
Methods A cross-sectional study was conducted in Cayenne Hospital from 1 January 2009 to 1 June 2014 in the hospital database. Only patients with the definition of the European Society of Cardiology (ESC) were included, based on the first transthoracic ultrasonography found during the study period. Medical files were consulted.
Results With 182 patients included, the prevalence of cardiomyopathies among heart diseases was estimated at 4.3% (95% CI 3.7% to 4.9%). Twelve patients had a familial or genetic aetiology (6.6%) and 170 a non-familial or non-genetic aetiology (93.4%). The morphological spectrum was: dilated for 114 patients (62.6%), hypertrophic for 27 (14.8%), unclassified for 1 (0.5%) and non-classifiable for 13 (7.1%). This group was constituted of patients with a systolic and/or diastolic functional impairment without morphological abnormality. The aetiological spectrum found 184 aetiologies including: 70.9% undetermined, 8.7% infectious (with 6.5% chagasic and 0.5% related with human immunodeficiency virus) and 6.0% with toxins.
Conclusions Cardiomyopthies are a common and severe clinical problem. The frequency of infectious aetiologies and dilated impairment are arguments for cardiomyopathies with tropical particularities. However, the preponderance of undetermined aetiologies justifies the development of further research programmes.
- cardiomyopathy dilated
- cardiomyopathy hypertrophic
- chagas' disease
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Key questions
What is already known about this subject?
Cardiomyopathies are a very heterogenous group of structural and functional abnormalities of the myocardium that cannot solely be explained by coronary disease or abnormal loading conditions. In developed countries, the presentation and natural history of cardiomyopathies originates from a small number of cohorts in specialised centres. In sub-Saharan Africa, the causes of cardiomyopathy are dominated by cardiomyopathies linked to the HIV virus infection, idiopathic dilated cardiomyopathies, endomyocardial fibrosis and hypertrophic cardiomyopathies. In Latin America, where Chagas disease is endemic, there are rare data on cardiomyopathies.
What does this study add?
The present provides the first data on cardiomyopathies in an Amazonian environment showing that for cardiomyopathies with a clear aetiological conclusion, infectious causes predominated in association with dilated cardiomyopathies of non-familial non-genetic cardiomyopathies. However, despite diagnostic progress, cardiomyopathies of undetermined aetiology remain the most frequent.
How might this impact on clinical practice?
Specific efforts should aim to better diagnose, characterise and treat this serious health issue.
Introduction
Cardiomyopathies are structural and functional abnormalities of the myocardium that cannot solely be explained by coronary disease or abnormal loading conditions. They are a very heterogenous group of disorders that represent a significant health burden by causing death from arrythmia, progressive failure and stroke. In developed countries, the presentation and natural history of cardiomyopathies originate from a small number of cohorts in specialised centres. In most low-income and middle-income tropical countries, the epidemiological transition from infections and nutritional deficiencies to chronic non-communicable diseases is well underway.1 Among the non-communicable causes of death cardiac failure concerns 37.7 million persons. In the absence of publications estimating the burden of cardiomyopathies, one way to try to estimate this burden is to infer cardiomyopathies from the causes of cardiac failure. The most frequent causes of cardiac failure in the world remain ischaemic heart disease, hypertensive cardiopathy, rheumatic heart disease and chronic obstructive disease. Cardiomyopathies could then be the next important cause on the list. The literature reports that in sub-Saharan Africa, the causes of cardiomyopathy are dominated by cardiomyopathies linked to the HIV virus infection, idiopathic dilated cardiomyopathies, endomyocardial fibrosis and hypertrophic cardiomyopathies.1 Publications are lacking to improve knowledge on the global burden of cardiomyopathies among cardiopathies. In Latin America, where wide socioeconomic disparities exist, cardiac failures represent an important part of hospitalisations, with a great variety of causes, among which idiopathic dilated cardiomyopathy represents 1.3%–37% of cardiac failure cases, with Chagas disease credited for 1.3%–21% of cardiac failure cases.2 In French Guiana, an overseas French territory in the Amazon basin, cardiovascular diseases represent the second cause of death.3 The mortality excess for cardiovascular diseases is +63%, with a lower mortality from ischaemic heart diseases than mainland France (−24.8%). However, mortality due to heart failure is much higher than in mainland France (+92.2%).4
Apart from a recent publication on a beriberi epidemic among illegal gold miners,5 a study on Pompe’s disease in infants of Maroon ethnicity6 and a study showing the high mortality of children aged less than 1 year due to cardiac malformations,7 there have been no other studies on the aetiological spectrum of cardiac failure and their poor prognosis in French Guiana. Infectious diseases may have an important role in this tropical context, but there may be other specificities.8 9 The present study aimed to answer the following question: what is the morphological and aetiological spectrum of cardiomyopathies in this French territory with an Amazonian biome?
The main objective of the study was thus to describe the morphological and aetiological spectrum of cardiomyopathies treated in Cayenne General hospital between 1 January 2009 and 1 June 2014. The secondary objectives were to estimate the prevalence of cardiomyopathies among cardiopathies in Cayenne General Hospital, to describe the observed morbidity and mortality of cardiomyopathies and to make comparisons with the literature.
Material and methods
Definitions
The definition of cardiomyopathy was: ‘myocardial anomaly where the myocardium is structurally and functionally abnormal, in the absence of coronary disease, hypertension, valvular pathology or congenital heart disease sufficient to cause the observed anomaly’.10 Cardiac failure was defined as: ‘clinical syndrome characterised by typical symptoms which can be accompanied by clinical signs caused by a structural and/or functional anomaly that reduces the cardiac output and/or increases the intracardiac pressures at rest or during stress’.11 Systolic cardiac failure, now called cardiac failure with a reduced ejection fraction, it is defined ultrasonographically by an ejection fraction of the left ventricule <40% associated with clinical symptoms and/or signs.11 Diastolic cardiac failure, now called heart failure with preserved ejection fraction (HfpEF), is defined by an ejection fraction of the left ventricule ≥50% with abnormal measures of the diastolic function and clinically the presence of symptoms and/or signs of cardiac failure.11 Cardiopathy: nosological grouping of the international classification of diseases codes of diseases and syndromes linked to a heart disease. Pericarditis: pericarditis was defined by cardiac ultrasonography identifying the presence of fluid in the pericardial space.
Type of study
The study was observational, cross-sectional, descriptive and monocentric. The study was conducted at Cayenne General Hospital between 1 January 2009 and 1 June 2014. The date of 1 January 2009 was chosen because prior to this the quality of the data was heterogenous. The end date of 1 June 2014 was chosen in order to have at least 2 years to observe any complications and to monitor survival of the last included patient.
Study population
The source population consisted of outpatients or inpatients identified by the medical information system of the hospital with an exit date between 1 January 2009 and 1 June 2014. The target population consisted of inpatients or outpatients with an exit diagnosis of cardiomyopathy according to the 2008 European Society of Cardiology (ESC) definition.
Jugdement criteria
The main jugdement criteria was the diagnosis of cardiomyopathy according to the 2008 ESC definition.10 Following the ESC 2008 definition, there is no mention of the systolic or diastolic dysfunction regarding functional abnormalities of the myocardium. Hence, we included all patients and then described functional dysfunction according to the transthoracic ultrasonography reports. Patients with myocarditis were not included. The secondary jugdement criteria were morbidity defined as a complication from a cardiomyopathy, mortality defined as a documented death, severity at the time of inclusion clinically defined as: chronic cardiac failure, dyspnoea greater than New York Heart Association (NYHA) grade 2 level and/or an ejection fraction <40% on inclusion.
Inclusion, non-inclusion and exclusion criteria
Inclusion criteria were patient seen at Cayenne General Hospital diagnosed with cardiomyopathy according to the ESC 2008 definition with an exit date between 1 January 2009 and 1 June 2014; patients were not included if they were not followed at Cayenne General Hospital; exclusion criteria were absent or non-available hospitalisation notes; verification of the medical files ruling out cardiomyopathy; and patient refusal.
Ethical and regulatory aspects
The database was declared to the Commission Nationale Informatique et Libertés (CNIL N° 1959321). For each included patient, as required by French regulations, we enquired whether the patient objected to us consulting the medical files using an individual phone call. When the patient was lost or had died, we considered that there was no opposition; as required by French law, we had attempted to contact patients to seek non-opposition.
Study conduct
Queries in the hospital medical information system using CoRaActivité software searched for the 10th International Classification of Diseases (ICD-10) codes for ‘diseases of the circulatory system’, a broad search as a main diagnosis or associated diagnosis. Among these, we tried to identify those with cardiomyopathies first using the ICD-10 codes for cardiomyopathy, then we tried to identify false negatives by searching for codes for some major complications of cardiomyopathies: cardiac failure, rhythmologic or conduction problems and cardiorespiratory arrest.
We also tried to identify false positives. Three groups of patients were identified: those coded cardiomyopathy; those coded as cardiac failure; and those coded as rythmological or conduction problems and/or cardiac arrest. For these three groups, all hospitalisation reports were consulted by the first author PL. After checking the hospitalisation summary report to eliminate false positives, medical files were consulted to further verify and to collect variables of interest. For unclassified or mixed cases, full medical records were reviewed with the senior cardiologist (second author KF).
Inclusion of patients: prevalent/incident cases
The moment of inclusion was defined by the first ultrasound identifying cardiomyopathy. This ultrasound was contemporary to the clinical event identified in our query in the medical information system. The date of the ultrasound was the reference date for the data collection in the medical records. Cardiomyopathies being mostly long-term diseases, the data were collected after the acute episode when possible. The acute episode corresponded either to a punctual complication or an inaugural passage to a more advanced stage of the disease. Incident cases were patients for whom the diagnosis was not known prior to the inclusion. If for one of these patients inclusion was performed during an acute episode, we considered the first transthoracic ultrasound beyond the acute episode within 3 months after the acute episode. Prevalent cases were patients for whom the diagnosis was known before the inclusion in our study. If for one of these patients inclusion was performed during an acute episode, we considered the first ultrasound within 3 months before the acute episode.
Data collection
Sociodemographic variables, comorbidities, risk factors and family history were collected using EpiData. Complications of cardiomyopathies at the time of and before inclusion were collected to identify early manifestations of cardiomyopathies. The circumstances leading to the diagnosis and the details of the ultrasound at inclusion were collected. To be reported in our study, the aetiology had to formally appear in the medical records. At the date of 1 June 2016, the mention of death was searched in the medical records or in the medical information system. In the absence of mention of death, we searched for the last visit inCoRa Document.
Statistical analysis
Anonymised data were analysed with Stata V.13.
The population was described with medians, quartiles, frequencies, means and SD where appropriate. Prevalence calculation was completed with 95% CIs.
Results
After verification in the information system and in the medical records, 182 patients were finaly retained for analysis (online supplementary file 1). The prevalence of cardiomyopathies among cardiopathies was 4.3% (95% CI 3.7 to 4.9).
Supplemental material
The yearly repartition of the number of inclusions was homogeneous ranging from 26 inclusions in 2006 to 38 in 2012. The average monthly number of cases was 2.8 (±0.4). The annual number of cases by aetiology was also homogeneous.
Prevalence of different morphological aspects of cardiomyopathies
Overall 62.6% of patients had dilated cardiomyopathy (n=114), 14.8% were hypertrophic (n=27), 14.8% were mixed morphology (n=27; 23 hypertrophic+dilated, 3 dilated+unclassified and 1 hypertrophic+unclassified) and 7.1% were labelled unclassifiable because there were no morphological anomalies, just functional impairment in the absence of other causes. Finally one patient was not classified, having a non-compaction of the left ventricule. There were no restrictive morphologies and no arythmogenic cardiomyopathy in our population.
Aetiological description according to the 2008 ESC classification
Table 1 shows the aetiological classification. Non-familial or non-genetic cardiomyopathies represented 93.4% of patients (n=170), whereas familial or genetic forms represented 6.6% of patients (n=12). Two patients had two concomitant non-genetic non-familial aetiologies.
Classification according to the adapted* 2008 European Society of Cardiology of cardiomyopathies observed using transthoracic ultrasonography: Cayenne between 1 January 2009 and 1 June 2014, French Guiana
Table 2 synthesises the cardiomyopathy aetiologies identified in 29.1% of patients.
Identification of familial and non-familial aetiologies of cardiomyopathies observed using transthoracic ultrasonography at Cayenne General Hospital between 1 January 2009 and 1 June 2014
Figure 1 shows the respective frequency of the 184 aetiologies identified. Globally, the main aetiologies identified were: undetermined aetiology (70.1%), infectious aetiology (8.7%), familial or genetic (6.5%), toxic (6.0%) and peripartum cardiomyopathies (3.3%). Other aetiologies were found in 4.9% of cases. This group included cardiomyopathies caused a range of aetiologies: autoimmune, endocrine, nutritional, postradiation, rythmic, surcharge, sequelae from myocarditis and a significant atheroma lesion in a mixed aetiology. One patient (0.5%) had idiopathic dilated cardiomyopathy.

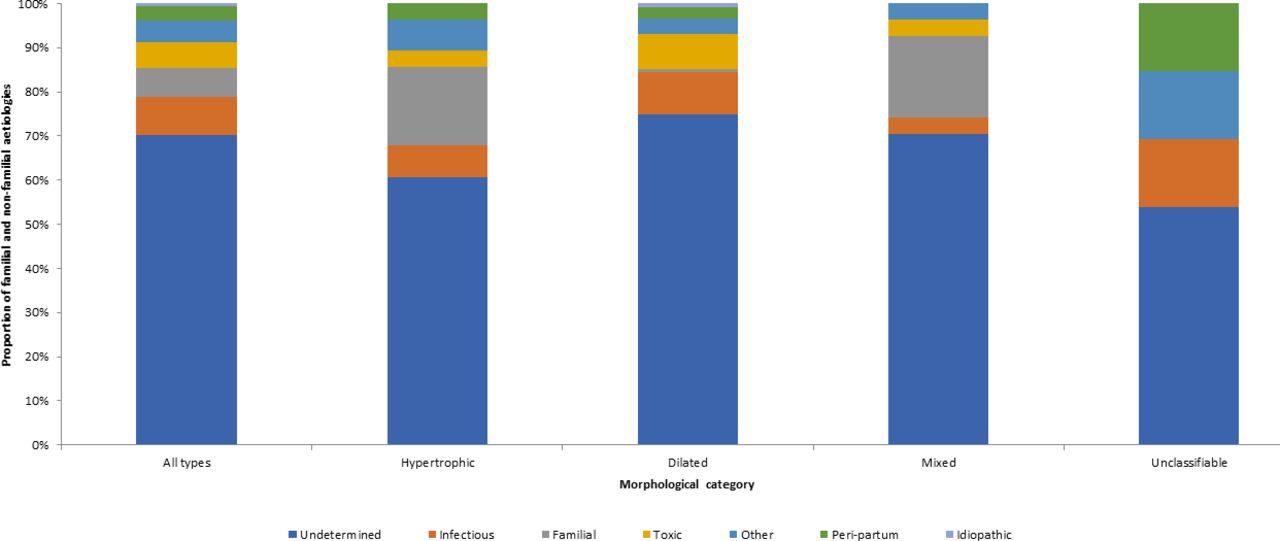
Proportion of cardiomyopathies of familial and non-familial aetiologies by ultrasonographic morphological class: Cayenne, French Guiana, 2009–2014.
Undetermined aetiologies were found for at least half of patients whatever the morphology. Infectious aetiologies were mostly found for dilated cardiomyopathy (9.6%) and unclassifiable morphologies (15.4%). Familial aetiologies were found in hypertrophic (18.5%) and mixed cardiomyopathies (18.5%). They were not observed in unclassifiable cardiomyopathies. Toxic aetiologies were mostly found in dilated cardiomyopathies (7.8%); they were not found in functional forms. Peripartum cardiomyopathies were found whatever the morphological or functional impairment and mostly as unclassifiable (15.4%).
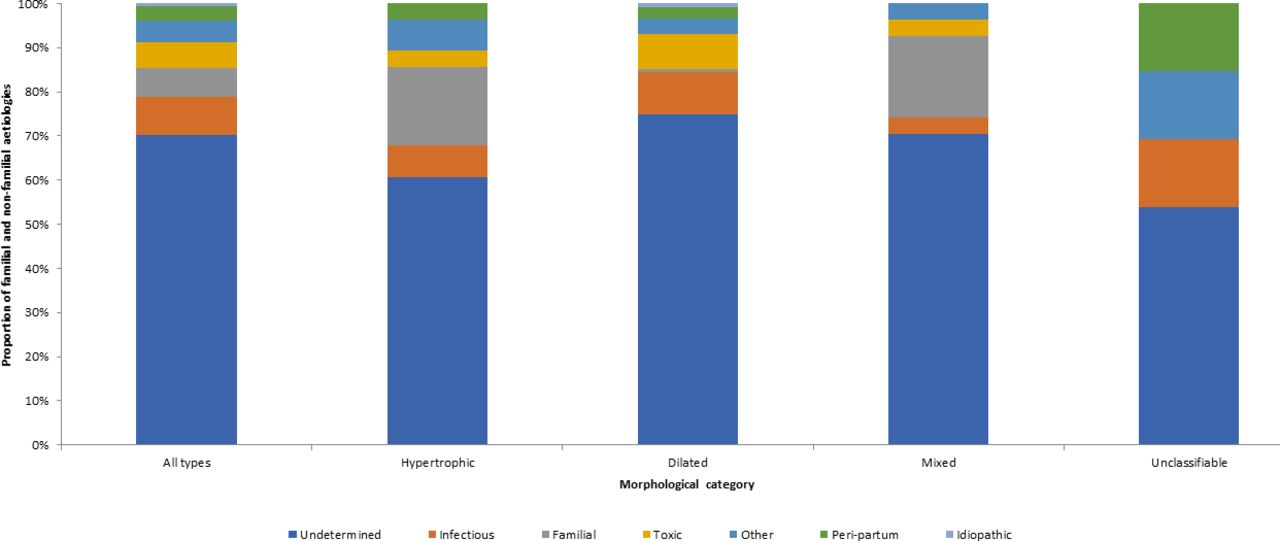
Figure 2 summarises the aetiologies identified in patients with non-familial non-genetic cardiomyopathy. For the 170 patients with non-familial non-genetic cardiomyopathies, the main aetiologies were undetermined (75%), infectious (9.3%), toxic (6.4%), peripartum cardiomyopathy (3.5%) and ‘other etiologies’. Undetermined aetiologies were found for both morphological and functional impairments. Infectious aetiologies were also found in morphological and functional anomalies; the two main causes were Chagas disease for 12 cases (7.0%) and HIV infection for 1 case (0.5%). Toxic aetiologies were found for dilated, hypertrophic ormixed cardiomyopathies. The main toxic aetiologies identified were chronic alcoholism (3.5%), medications (2.3%) and drug use (0.6%). Peripartum cardiomyopathies included both morphological and functional impairment.
{kind=link}
{kind=link}
Proportion of cardiomyopathies of non-familial aetiologies by ultrasonographic morphological category: Cayenne, French Guiana, 2009–2014.
Genetic and familial cardiomyopathies
The main genetic/familial aetiologies found were left ventricule non-compaction for 5/12 patients (2.7%) and Pompe’s disease for 3/12 patients (1.6%). Others were single cases of Noonan syndrome, protein C gene mutation and cardiofaciocutaneous syndrome.
Study population
The sex ratio was 2.6 males per female. The mean age at inclusion was 51.6 years. At the time of inclusion, the diagnosis of cardiomyopathy was known for 25.8% of patients (prevalent cases), whereas for the rest (74.2%), the diagnosis was unknown (incident cases). The mean age at diagnosis was 49.9 years (range 0–93). Overall, at the time of diagnosis, 45.6% of cardiomyopathy cases were aged 41–60 years. Most patients were from French Guiana (45.1%) and the rest from the Guiana Shield (22.5%). Most resided on the coastal area (84.1%).
A family history of cardiomyopathy was found in 11.0% of patients. Nearly half of patients (49.0%) had comorbidities on admission. The most common were chronic alcoholism (11.5%), nephropathy (9.3%) and cancer (7.7%). Cardiovascular risk factors were found in 65.9% of patients, mostly hypertension (48.9%) and a history of smoking (24.7%).
Clinical characteristics are presented table 3. Briefly, a third of patients were at the cardiac failure stage when included (33.0%). This proportion was lower in the unclassifiable group (7.7%). Nearly half of patients (46.7%) had an NYHA score >2 for both functional and morphological anomalies. The proportion of patients with a NYHA score >2 was multiplied by 4 between patients included outside of an acute episode (13.5%) and those included during an acute episode (55.2%).
Clinical, paraclinical and therapeutic characteristics of cardiomyopathy cases observed with transthoracic ultrasonography at Cayenne General Hospital between 1 January 2009 and 1 June 2014
An acute clinical presentation was found for most patients, whether impairment was morphological or functional (79.7%). The clinical situations encountered were: acute cardiac failure (64.8%); cardiac rhythm or conduction anomalies (31.3%); more frequent in the unclassifiable functional impairment (61.5%); and chest pain, lipothymia/loss of consciousness and stroke (2.8%).
Nearly a third (31.9%) of patients had previously presented a complication of their cardiomyopathy before inclusion in the study: acute cardiac failure (20.3%), rhythm or conduction anomalies (17.0%), stroke (3.9%) and heart transplant (0.5%).
Table 3 shows paraclinical and therapeutic characteristics of the study population. Reported ejection fractions are reported globally, independently from the clinical presentation. The median ejection fraction was 35%. Most patients (73.5%) had an ejection fraction <40% at the time of inclusion, notably dilated and unclassifiable forms (87.7% and 77%, respectively). There were no significant differences in the proportion of patients with an ejection fraction <40% whether the patient was included during an acute episode or outside of an acute episode (64,9% and 73,1%, respectively; data not shown). Systolic cardiac failure was the most common ultrasonographic finding (67.6%), notably in dilated and unclassifiable forms (78.1% and 77%, respectively). Diastolic cardiac failure was found in 3.3% of cases (never in the dilated category and 23% in the unclassifiable).
Half of patients had ongoing treatment at the time of inclusion (50.0%).
Description of ultrasonographic findings on inclusion
The main transthoracic ultrasound findings are presented in table 4. Kinetic anomalies were frequent (89%). Global hypokinesia was reported in 46.2% of patients, notably in dilated (53.5%) and unclassifiable forms (46.1%). Associated valvulopathies were reported for 79.1% of patients.
Main ultrasonographic findings for cardiomyopathies observed with transthoracic ultrasonography in Cayenne General Hospital ultrasonographic data of cardiomyopathies at Cayenne General Hospital between 1 January 2009 and 1 June 2014
Circumstances of diagnosis
Most patients were symptomatic when diagnosed (78.6%). The main clinical elements reported were acute cardiac failure symptoms, rythm or conduction anomalies (28.6%) notably in the unclassifiable forms (53.9%) and chronic cardiac failure symptoms (24.7%). These elements were not available for 12.8% of patients. Among asymptomatic patients (9.3%), only one (0.5%) was diagnosed in the context of a family history, while the others were fortuitous discoveries (8.8%).
Patient evolution
Of the 182 patients, 22 patients were confirmed dead (12.1%), 5/27 (18.5%) with hypertrophic cardiomyopathy, 9/114 (9.6%) with dilated cardiomyopathy and 6/27 (22.2%) with mixed cardiomyopathy. The median time to death was 4 years (IQR 2–6 years).
Discussion
With a prevalence of 4.3% among cardiopathies (95% CI 3.7% to 4.9%), cardiomyopathies were relatively frequent. This represented 1/20 patients seen for a cardiological motive in our hospital and three patients every month. Overall, the results suggest that, despite probable under-recognition, the burden of cardiomyopathies in French Guiana is not negligible and that patients are diagnosed late. Hence, the advanced age at inclusion and the severe symptomatic profile suggest that there is limited awareness in the medical community and that referrals are late. Cardiomyopathies mainly concerned middle aged men, except for familial or genetic cardiomyopathies, which concerned younger patients.
The observed morbidity was important with a third of patients at the stage of chronic cardiac failure, nearly half with NYHA >2 and two-thirds of patients with an ejection fraction <40%. However, 79.7% of our patients were included outside of an acute episode. In acute situations, dyspnoea does not necessarily represent the basal state of the patient. Thus, the proportion of patients with a NYHA >2 increased from 13.5% outside of an acute episode to 55.2% during an acute episode. In addition, this score has the limitation of being subjective, notably when evaluating cardiomyopathies.12 In contrast with the NYHA score, the proportion of patients with an ejection fraction <40% was similar between those included during (64.9%) or outside and acute episode (73.1%). The measure of the ejection fraction is known to be an imperfect reflection of systolic function in hypertrophic cardiomyopathies. It only drops at a very advanced stage of the disease and for patients who do not have any ejection obstruction.13
The observed case fatality was significant (12.1%) despite the short observation period of our study and presumably incomplete reporting. The median delay of death was relatively short at 4 years.
In our study only two patients (1.1%) benefited from a heart transplant and none presented a cardiorespiratory arrest. It is commonly admitted that cardiomyopathies are known causes of cardiac arrest; therefore, it is likely that in patients with cardiac arrest, the lack of in-depth investigations after sudden death could explain this.14
The most frequent morphology observed was dilated cardiomyopathies (62.6%), followed by hypertrophic (14.8%), mixed (14.8%) and unclassifiable (7.1%). In the subgroup of familial and genetic cardiomyopathies, this trend was reversed: 41.6% of patients had a hypertrophic cardiomyopathy and 8.3% a dilated cardiomyopathy.
This trend was similar in the European registry of familial cardiomyopathies.15 American16 and Australian17 studies, in paediatric populations, found dilated cardiomyopathies before hypertrophic cardiomyopathies. However, these studies were not restricted to familial or genetic cardiomyopathies, and myocarditis was included. In Finland, a study on idiopathic paediatric cardiomyopathies also found a predominance of dilated cardiomyopathies.18
The literature review on cardiomyopathies did not allow comparisons because they were mostly focused on the aetiological spectrum of cardiac failure or the morphological spectrum of a given aetiology, or in particular population subgroups. Only the European registry of familial cardiomyopathies allowed some comparisons: the median age of patients with familial cardiomyopathy in our study was younger than in Europe (17 vs 42 years).19 A majority of patients were symptomatic at the time of diagnosis in both studies; fortuitous diagnosis or targetted familial screening was rare in both studies. In our study, the clinical status seemed more severe than what was reported in the European registry: 41.7% with NYHA >2 vs 20.4%, respectively.
In French Guiana, familial or genetic forms affected 6.6% of the study population. The main diagnosis in this group was non-compaction of the left ventricule (42% of familial forms in our study vs 3.2% in the European registry). Apart from the small sample size, this high frequency may reflect the fact that the diagnosis is based on ultrasonographic criteria, not genetic criteria, and thus is easier to detect. Pompe’s disease was observed in three patients in our study and in none in the European registry. A recent study showed the very high prevalence of heterozygocy among Maroon mothers in western French Guiana. Noonan syndrome was observed once in our study and once in the European registry (0.1%). Hypertrophic familial cardiomyopathy, which represents 1/500 births in the general population, was not observed in French Guiana.20 With 6591 births in French Guiana in 2014,21 one could expect 13 affected children, each year. The initial lack of symptoms, the rarity of genetic investigations and the unfamiliarity of physicians with this disease could explain the absence of identified cases during the study period.
Among non-familial non-genetic cardiomyopathies, excluding the undetermined aetiologies, infectious causes (8.7%) and toxic causes (6%) were notable. The importance of infectious diseases, notably Chagas disease, is not surprising in French Guiana. Chagasic cardiomyopathy may explain 8.1%–41% of cases of cardiac failure in Latin America.22 23 With six cases, peripartum cardiomyopathy was the third cause in non-familial aetiologies with a similar incidence than in Martinique.24 No cases of endomyocardic fibrosis were observed. This type of cardiomyopathy has been observed in Brazil, Colombia and Venezuela.25 26 The polymorphous nature of the disease and the difficulty to establish the diagnosis could explain this absence.
It was surprising to find no case of vitamin B1 deficiency (beriberi) since an ongoing epidemic among illegal gold miners was described in the interior of French Guiana in 2013.5 The management of these patients in remote health centres or the improper coding of disease could explain this absence.
The present study has significant limitations. The sample size in this single centre was relatively small, and diagnosis and follow-up were assessed with simple diagnostic tools. Of particular concern, the small proportion of non-familial forms was likely to be grossly underestimated, particularly for HCM and HCM-mixed phenotypes. This result coupled with the great proportion of patients with undetermined aetiology emphasises the unmet need for more adequate diagnostic work-up in patients with cardiomyopathies in the region. The cross-sectional, retrospective, monocentric design may have introduced different biases. The constitution of the database may have led to selection bias due to possible heterogeneities in the data quality.27 Moreover the ICD-10 is used mostly used for billing purposes and is different from the clinical classifications used. Recoding required making choices but this was sometimes difficult, given the ESC definition requirements, when reading the hospitalisation reports. Cayenne General Hospital is the only cardiology centre of the territory, which may have led to recruitment bias. Cases only seen in private practice or in the two other hospitals in French Guiana may have led to prevalence underestimation. In addition, because Cayenne General Hospital is the only cardiological centre, this possibly biased estimates towards greater severity. Finally, ultrasonographic results and medical conclusions in the medical files may have introduced variations depending on the physicians involved.
Despite these limitations, the present study has some strengths. To our knowledge, it is the first to evaluate all cardiomyopathies using the ESC classification, notably allowing the description of the morphological and aetiological spectrum of non-familial forms. Indeed, different registries only focus on certain types of cardiomyopathies or certain population groups. Second, this study illustrates the difficulty represented by the nosological concept of cardiomyopathy. It is easy to get lost in the literature where there is an abundance of different definitions and classifications, without real consensus,12 and used in alternation between studies. This is compounded by the practical difficulty of a morphological or aetiological diagnosis. In its definition, the ESC tacitly leaves the clinician decide the part attributed to the observed anomalies of an eventual association with hypertension, coronaropathies, valvulopathies or a congenital cardiopathy. It is only if this attributed part is deemed not important that the diagnosis of cardiomyopathy can be proposed. The contribution of complementary explorations allows ruling out many competing hypotheses. The difficulty of performing certain investigations and the absence of multidisciplinary synthesis may have led to a high frequency of undetermined aetiologies in our study (70.9%).
Third, the present study allows examining the concept of tropical cardiomyopathy put forward in 1967 and questioned in 2013.28 29 In French Guiana, although the epidemiological transition has largely occurred, infectious diseases still weigh on populations and largely affect the spectrum of cardiomyopathies, arriving as the first cause of non-genetic non-familial cardiomyopathies with an identified cause. Hence, if the morphological spectrum differs between tropical and non-tropical areas, this is presumably increasingly attenuated since 1967 by human migrations and development. The nebulous entity of cardiomyopathy of unknown origin described by Shaper28 has been clarified by the progress of diagnostic investigations. In 1967, it was supposed that there was a specific factor that was restricted to tropical regions. From now on, precise aetiologies may be identified without being strictly restricted to tropical areas. Cardiomyopathies in tropical regions reflect local epidemiological conditions, which are often incomplete, with undetermined causes still leading the aetiological spectrum.
Conclusion
The present study suggests that, despite probable under-recognition, the cardiomyopathy burden in French Guiana is not negligible and that patients are diagnosed late. The advanced age at inclusion and the severe symptomatic profile suggest that there is limited awarenesss in the medical community and that referrals are late. The premise of this work was to attempt to test the hypothesis that the Amazonian biome would change the morphological and aetiological spectrum of cardiomyopathies in French Guiana. Despite a number of limitations, the present study showed that for cardiomyopathies with a clear aetiological conclusion, infectious causes predominated in association with dilated cardiomyopathies of non-familial non-genetic cardiomyopathies. For familial cardiomyopathies, hypertrophic forms predominated. However, despite diagnostic progress, cardiomyopathies of undetermined aetiology remain the most frequent emphasising that diagnostic work-up should be improved before concluding about the differences in the aetiological profile of non-infectious cardiomyopathies between French Guiana and the USA or Europe. The medical community of French Guiana should further research this problem in order to diagnose and treat this serious health issue earlier.
References
Footnotes
Contributors PL and AA planned the study, PL, NB, FC and KF conducted the study; PL wrote the initial draft; AA and MN edited the initial draft. AA is the article guarantor.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests None declared.
Patient consent for publication Not required.
Ethics approval Hospital Ethical committee (n°1–2016 V1) and Commission Nationale Informatique et Libertés (CNIL N° 1959321). The study was retrospective. For each included patient, as required by French regulations, we enquired whether the patient objected to us consulting the medical files using an individual phone call. When the patient was lost to follow-up or had died, we considered that there was no opposition, as required by French law we had attempted to contact patients to seek non-opposition.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Data are available on reasonable request. Data may be requested at cicec@ch-cayenne.fr. Prior to this, clearance to transfer data must be obtained from the commission nationale informatique et libertés 3 Place de Fontenoy - TSA 80715 - 75334 PARIS CEDEX 07.