Article Text

Abstract
Background Mutations in genes encoding ion channels or sarcomeric proteins are an important cause of hereditary cardiac disease. However, the severity of the resultant disease varies considerably even among those with an identical mutation. Such clinical variation is often thought to be explained largely by differences in genetic background or ‘modifier genes’. We aimed to test the prediction that identical genetic backgrounds result in largely similar clinical expression of a cardiac disease causing mutation, by studying the clinical expression of mutations causing cardiac disease in monozygotic twins.
Methods We compared first available clinical information on 46 monozygotic twin pairs and 59 control pairs that had either a hereditary cardiomyopathy or channelopathy.
Results Despite limited power of this study, we found significant heritability for corrected QT interval (QTc) in long QT syndrome (LQTS). We could not detect significant heritability for structural traits, but found a significant environmental effect on thickness of the interventricular septum in hypertrophic cardiomyopathy.
Conclusions Our study confirms previously found robust heritability for electrical traits like QTc in LQTS, and adds information on low or lacking heritability for structural traits in heritable cardiomyopathies. This may steer the search for genetic modifiers in heritable cardiac disease.
- Genetics
- Twin Study
- Cardiomyopathy
- Channelopathy
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Key messages
What is already known about this subject?
Mutations in genes encoding ion channels or sarcomeric proteins are an important cause of hereditary cardiac disease. However, the severity of the resultant disease varies considerably even among those with an identical mutation. This may be caused by genetic or environmental factors.
What does this study add?
This study confirms previously found robust heritability for electrical traits like corrected QT interval in long QT syndrome and adds that heritability for structural traits in heritable cardiomyopathies is likely low.
How might this impact on clinical practice?
The results of this study will probably not alter clinical practice, but do provide information that may steer the search for genetic modifiers in heritable cardiac disease.
Introduction
A number of hereditary cardiac diseases are caused by autosomal dominantly inherited, single gene mutations.1 2 Still, the extent and severity of the main cardiac abnormality caused by such mutations varies considerably between mutation positive subjects. Causes for this variability are largely unknown. It is widely speculated that this variability may be caused by variation in genes other than the mutated one, for instance by altering the clinical expression of the disease-causing mutation.3 This assumption predicts that two carriers of such a mutation who have an identical genome should have quite similar clinical expression of the mutation they both carry. Based on this idea, it is expected that recently developed high-throughput technologies may identify additional genetic variants that modify the cardiomyopathy or channelopathy.4
However, it is unclear whether this expectation is justified, as it has not yet been tested to what extent identical genomes indeed result in a comparable clinical expression of a disease-causing mutation. Here, we tested this by analysing whether mutation-positive monozygotic twins are more concordant for the main disease trait than mutation-positive dizygotic twins or siblings. We analysed clinical data from twins counselled in high-volume cardiogenetic centres around the world and added data from twins on whom data were available publicly.
Monozygotic twins illuminate the genomic contribution to complex diseases.5–7 Twin studies are based on the assumptions that (1) monozygotic twin pairs share 100% of their genome, and therefore share additive genetic effects (the added effect at a single locus of one allele added to the effect of the other allele) and dominance effects (effects of interaction between two alleles on the same locus); (2) dizygotic twin pairs and normal siblings share 50% of their genome, so they share 50% of the variance in additive genetic effects and 25% of the variance in dominance effects8; (3) monozygotic and dizygotic twin pairs share common environmental effects to the same extent.9 10 For this study, we assumed that non-twin siblings share common environmental effects to the same extent as twin siblings. Although it is well known that even monozygotic twins do not have identical genomes,11 the current analysis allows to estimate the probability to find modifiers based on high-throughput genomic sequencing.
Methods
We collected monozygotic twin pairs with dilated cardiomyopathy (DCM), hypertrophic cardiomyopathy (HCM) or long QT syndrome type 1, 2 or 3 (LQTS) from families with a high suspicion of familial disease in which a (likely) pathogenic mutation was identified. In total, we recruited 36 monozygotic twin pairs from Amsterdam and Groningen (The Netherlands), Rochester New York and Rochester Minnesota (USA), Helsinki (Finland), Pavia and Firenze (Italy), Umea (Sweden), Copenhagen (Denmark), Paris (France) and Murcia (Spain). In all cases, monozygosity was established on clinical grounds, while in 27 pairs (75%), monozygosity was confirmed using polymorphic DNA markers. In addition, we added detailed clinical data on 10 pairs of monozygotic twins with a cardiomyopathy or LQTS, in which a pathogenic mutation was identified or inherited disease was strongly suspected, from previously published studies.7 12–21 Taken together, we collected data on 46 monozygotic twin pairs (table 1). As a control group, we collected data on 59 pairs of same-sex dizygotic twins or same-sex normal siblings (table 1). We included only same-sex control pairs to avoid sex-induced within-pair differences. These cases were selected from families in which one of the aforementioned hereditary cardiac diseases was diagnosed. Only retrospectively collected data were used in an anonymised database, which is in line with the guidelines for retrospective data analysis of the Amsterdam UMC institutional review board.
Monozygotic twin pairs and control pairs of which data were available for this study per disease
We retrospectively collected first available data on left ventricular ejection fraction (LVEF) and left ventricular end-diastolic dimension (LVEDD) in DCM, diastolic interventricular septum thickness (IVSd) in HCM and corrected QT interval (QTc, calculated using Bazett’s formula) for LQTS QTc (online supplementary table S1). We obtained values taken from the first available examinations, where the time between the measurements of two individuals within a pair did not exceed 10% of the age of the oldest individual. An exception to this were seven pairs (six of which were control pairs) where early data were available only for the more severely affected individual, while the less affected individual was only studied at a later age. For 13 LQTS control pairs, individuals were tested for the first time at different ages in the context of family counselling (online supplementary table S2).
Supplemental material
We found no differences in age or sex between the monozygotic twins and the control group using Mann-Whitney (two-tailed) U tests (table 2). As all pairs are sex-matched and the heritability estimations are performed using the relation between two members of a pair, and the tests were performed at the same age for both members of a pair, we did not correct for age or sex differences before performing the heritability estimations.
Baseline characteristics for the monozygotic twin pairs and control pairs per disease
To calculate the genetic contribution to a trait, we used structural equations modelling (SEM). When assessing the heritability of traits, the phenotypic variance can be split into genetic and environmental components and further divided into four components: A (additive genetic effects, which is narrow-sense heritability h2), D (dominance effects), C (common environmental effects) and E (environmental effects unique to the individual). ACE (additive genetic effects, common and unique environmental effects), AE (additive genetic effects and unique environmental effects) and CE (common and unique environmental effects) models were fitted and assessed for goodness of fit using the Akaike Information Criterion (AIC) where a low AIC indicates a good fit.8
Results
We compared first available clinical information on disease traits in 46 monozygotic twin pairs and in 59 control pairs of same-sex dizygotic twins or same-sex normal siblings (table 1). We found no differences in age or sex between the monozygotic twins and the control group (table 2). The results of the narrow-sense trait heritability (h2) estimations are summarised in figure 1 and table 3. SEM yielded an h2 of 4.9% (95% CI 0.0% to 84.5%) for LVEF and 52.8% (95% CI 0.0% to 91.2%) for LVEDD in DCM, 0.0% for IVSd (95% CI 0.0% to 43.6%) in HCM and 57.5% for QTc values in LQTS (95% CI 5.5% to 87.5%).
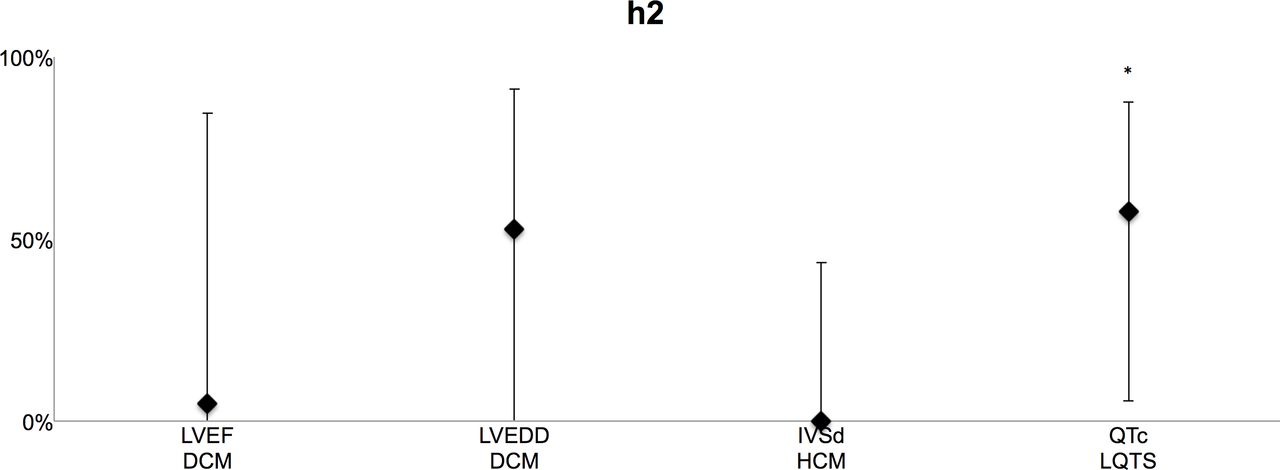
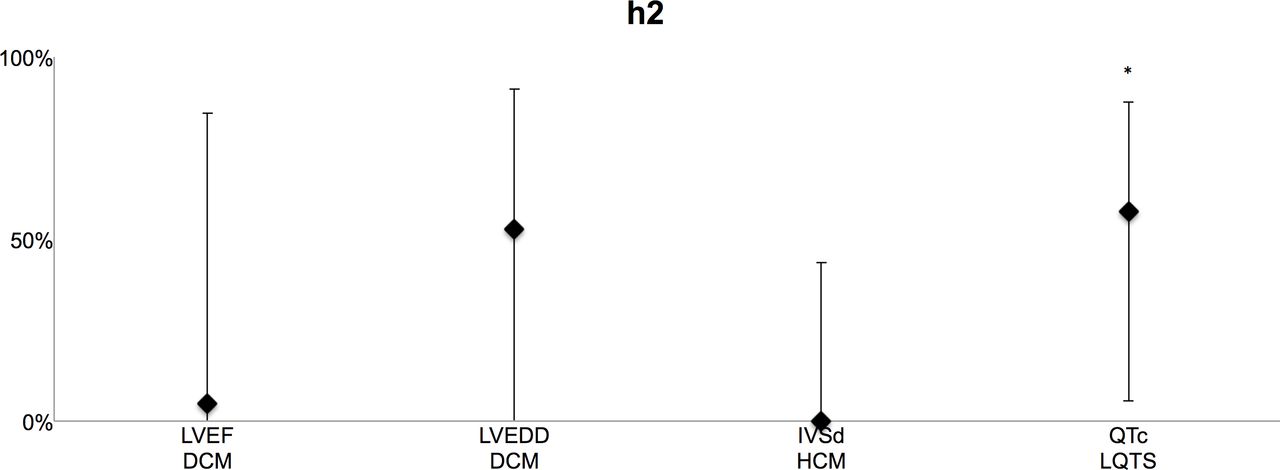{kind=link}
Heritability estimates in percentages using structural equations modelling for primary trait phenotypes in DCM (LVEF and LVEDD), HCM (IVSd) and LQTS (QTc). *Significant (within 95% CI). DCM, dilated cardiomyopathy; HCM, hypertrophic cardiomyopathy; IVSd, diastolic interventricular septum thickness; LQTS, long QT syndrome; LVEDD, left ventricular end-diastolic dimension; LVEF, left ventricular ejection fraction; QTc, corrected QT interval.
Heritability estimates as estimated using structural equations modeling (SEM)
For QTc values in LQTS, a model describing variation with additive genetic effects (AE model) had the best fit (p=0.03) with the observed data. In HCM, a model describing IVSd variation with shared environmental effects (CE model; p=0.02) had the best fit, and the contribution of shared environmental effects on IVSd variation was significant (65.6%, 95% CI 20.1% to 83.4%).
Discussion
In this study, we tested our hypothesis that monozygotic twins with a mutation that causes either LQTS or a cardiomyopathy are more concordant for the main disease trait than dizygotic twins or siblings that also carry such a mutation. Indeed, we found significantly higher concordance for QTc in monozygotic twins with a long QT causing mutation. However, we did not find such high or significant concordance for the primary cardiomyopathy trait of either LVEF or dilatation in DCM, or septal thickening in HCM.
In this worldwide collaboration, we collected a higher number of twins with a mutation that causes a cardiomyopathy or channelopathy than to our knowledge has been reported previously. We believe that there are probably not many more (monozygotic twins) out there as this collection is the result of a search in the cohorts collected by all major groups active in this field. Although this enabled us to study a relatively large number of twins, these particular twins are rare and the absolute numbers are still low.
Given the sample sizes in this study, varying from 19 to 55 twin pairs, at a significance level of p value <0.05, narrow-sense heritabilities of >0.80, 0.80, 0.60 and 0.55 could be found with 80% power for LVEF, LVEDD, IVSd and QTc, respectively. The power calculations were done with functions from Verhulst,22 which use the observed twin pair numbers and assume a common variance equal or smaller than the observed common variance. This suggests the heritability of QTc lengthening in LQTS we found is indeed significant. Our findings in LQTS confirm findings from other studies on the heritability of the QT interval trait.23–30
Our study cannot exclude that there is some heritability for structural traits in mutation-induced cardiomyopathies like HCM or DCM, and indeed for LVEDD in DCM our results show a trend for considerable heritability. However, it does suggest that heritability is less robust in these diseases, and that in these diseases a greater influence of environmental factors on the development of the disease trait(s) may be expected. Indeed, for the main trait of HCM, thickness of the cardiac septum, we even found a statistically significant contribution of environmental effects. The narrow-sense heritability of zero and the significant role for the environment on expression of septal wall thickness in HCM was unexpected since singular reports on monozygotic twins report both discordant14 and concordant twins.15
A downside of the use of data from multiple centres is that results may be affected by differing clinical standards and interobserver variability. This argument also applies to the use of detailed clinical data from clinical publications on twin pairs with HCM, DCM and LQTS. Because ratios within twin pairs have been used for the comparison with other twin pairs, and the two members of each pair were observed by the same clinician, we feel that interobserver variability does not influence our results. Furthermore, the members of each pair were observed around the same time, which both minimises intraobserver variability as well as age-dependent individual factors that may influence disease trait development. Finally, one may argue that the use of data from clinical publications could induce an inclusion bias. However, literature describes both concordant and discordant phenotypes, suggesting a bias by preferred publication of either type was not introduced.
In an effort to increase the number of cases in this study, we used dizygotic twin pairs as controls and also normal siblings. Both dizygotic twin pairs and normal siblings share 50% of their genome. We did not have information on the extent of shared environment for the studied cases, and we cannot exclude that this has introduced a bias in disease groups where normal siblings make up most, or even all of the controls. To minimise age-related and sex-related differences, we only selected same-sex control pairs and used data from comparable time points for all groups.
Many acquired differences can account for the discordant phenotypes we find in monozygotic twin pairs that share a disease-causing mutation, including post-translational modifications or environmental factors. Since we did not find larger differences between older twin pairs than younger pairs, it seems that lifetime exposure to environmental factors is not the only explanation for this discordance. Early acquired factors may already play an important role, as may de novo mutations, copy number variations31 and differences in maternally inherited mitochondrial DNA which varies even within an individual.32
Conclusion
We found a significant effect of genetic background on the clinical expression of mutations that cause LQTS, but did not find such effects for mutations that cause DCM and HCM. This suggests that environmental factors may be of relatively greater importance to explain differences in disease severity in families with a mutation causing DCM or HCM. Accordingly, it is anticipated that there are more genetic modifiers that underlie incomplete penetrance and variable expression of disease genes in LQTS, and that studies looking for genetic modifiers in heritable heart disease should focus more on disease with primary electrical phenotype like LQTS than disease with primary structural phenotype like DCM or HCM.
References
Footnotes
Correction notice This article has been corrected since it first published online. The open access licence type has been amended.
Contributors Planning: JAJ, KYvS-Z, AAMW, YMP. Conduct: JAJ, KYvS-Z, MWTT, JPvT, IC, JvdS, AMCV, JMB, AJM, HS, SGP, AR, JT-H, MJA, IO, PC, JRG, MPvdB, AAMW, YMP. Reporting: JAJ, KYvS-Z, MWTT, JPvT, MJA, AAMW, YMP. Overall: JAJ, KYvS-Z, YMP.
Funding This work was supported by the Netherlands CardioVascular Research Initiative (Project PREDICT and ARENA): the Dutch Heart Foundation, Dutch Federation of University Medical Centres, the Netherlands Organisation for Health Research and Development, the Royal Netherlands Academy of Sciences, the Windland Smith Rice Comprehensive Sudden Cardiac Death Program, the Finnish Foundation for Cardiovascular Research and the Novo Nordic Foundation.
Competing interests MA is a consultant for Boston Scientific, Gilead Sciences, Medtronic and St. Jude Medical. MJA and Mayo Clinic received sales-based royalties from Transgenomic for their FAMILION-LQTS and FAMILION-CPVT genetic tests. However, these entities had no involvement with this study. The remaining authors have nothing to disclose.
Patient consent for publication Not required.
Provenance and peer review Not commissioned; externally peer reviewed.