Article Text

Statistics from Altmetric.com
A number of prospective studies, dating back to the 1980s, have found that lower levels of plasma cholesterol correlate with increased risk for cancer; in some of these studies, it was noted that this correlation was strongest for cancers diagnosed within a year of cholesterol measurement, suggesting the possibility of reverse causality.1–11 More recently, Ravnskov and colleagues conducted a systematic review of cohort studies enrolling subjects over 60 in which the association of low-density lipoprotein cholesterol (LDL-C) with all-cause mortality and/or cardiovascular mortality had been evaluated.12 In 14 of the 30 total cohorts involved, LDL-C was inversely and significantly associated with all-cause mortality; in the remaining 16, no association was found. With respect to cardiovascular mortality, which was determined in nine cohorts, the association of LDL-C was inverse and significant in two and null in seven. The failure to find a positive a correlation between LDL-C and cardiovascular mortality is consistent with the previous research reporting that the positive correlation of LDL cholesterol and cardiovascular mortality that is clearly seen among younger subjects is substantially blunted among the elderly.
These findings have led some to suspect that low LDL-C plays a pathogenic role in cancer and perhaps other non-cardiovascular pathologies and that LDL-C is of little pathogenic significance for the progression atherogenic disease in the elderly.12 This interpretation evidently casts doubt on the wisdom of medicating the elderly for LDL-C control.
Counterarguments dispute the pathogenicity of low LDL
The thesis that low LDL-C may play a causative role in higher non-cardiovascular mortality has been challenged cogently by several lines of evidence. Law and colleagues13 compared cohort studies of groups of employees, most of whom could be presumed to be healthy at baseline, with community cohorts, which included many individuals with prevalent disease. They found that low total cholesterol in the employee cohorts was not associated with increased risk for non-cardiovascular mortality, whereas such an association was found in the community cohorts.13 They concluded that when low cholesterol correlated with increased risk in the community cohorts, this likely reflected reverse causation—an incipient disease state was responsible for a decline in plasma cholesterol. They did, however, confirm—as many others have14–18—that low cholesterol is associated with increased risk for haemorrhagic stroke in those with elevated blood pressure; presumably, LDL impacts the risk for cerebral aneurysms, or the fragility of these aneurysms. Nonetheless, the protection from ischaemic vascular disease associated with low LDL-C more than outweighed the adverse impact on haemorrhagic stroke risk in these Western cohorts.
Iribarren and colleagues examined data from the prospective Honolulu Heart Study, segregating the subjects into groups according to whether their plasma cholesterol rose, fell or remained stable during the first 6 years of follow-up.19 They found that subjects with low cholesterol at baseline, whose cholesterol remained low during follow-up, experienced no increase in risk for non-cardiovascular mortality as compared with those with average cholesterol. However, individuals whose cholesterol fell from mid-range to low during the follow-up period did experience an increased risk for cancer death and for non-cardiovascular/non-cancer death. It should be noted that, inasmuch as this study examined cholesterol changes over the years 1965–1974, most of the reductions in cholesterol observed likely were spontaneous, as opposed to being achieved by hypolipidemic medication.
The fear that statin therapy might increase cancer risk by lowering LDL-C has been put to rest by the outcomes of placebo-controlled clinical trials with statins. A meta-analysis of 27 statin treatment trials confirmed the expected reductions in major vascular events with statin therapy, while failing to note an effect of such therapy on risk for new cancer incidence or non-vascular mortality.20 A significant reduction of all-cause mortality was seen in patients who had no history of vascular disease at baseline.
Of particular interest is a meta-analysis of randomised trials comparing statins with placebos in the management of hyperlipidemic elderly subjects (over 65 years of age).21 This found that, in the statin-treated group, risk for myocardial infarction and stroke was significantly reduced, a trend towards a reduction in all-cause death did not achieve statistical significance (RR=0.941, 95% CI 0.856 to 1.035), and incidence of new cancer was not influenced. These findings evidently refute the notion that LDL-C loses its pathogenicity for atherosclerotic progression in the elderly.
Perhaps the most conclusive evidence disputing a causal relationship between low LDL-C and cancer is provided by a Mendelian randomisation study examining the association between genetic polymorphisms associated with lower LDL-C and cancer risk. Importantly, although low LDL-C was strongly associated with increased cancer risk in the cohorts examined, none of the genotypes linked to lower LDL-C—some associated with LDL-C reductions as great as 38%—were associated with higher cancer risk.22 In marked contrast, Mendelian randomisation studies have concluded that elevated LDL-C has an even stronger impact on risk for cardiovascular disease than that reported in observational epidemiology (presumably reflecting the fact that a single measurement of LDL-C is a rather imprecise gauge of life-long LDL-C exposure).23 24 Mendelian randomisation likewise confirms that LDL-C remains a mediating risk factor for vascular disease in the oldest old.25
It may also be noted that risks for so-called ‘Western’ cancers have been found to be comparatively quite low in quasi-vegan societies characterised by very low LDL-C levels.26 This clearly speaks against low LDL-C playing an important role in the induction of such cancers. Moreover, coronary disease is a minor cause of death in such cultures.
Online supplementary Table 1 summarises the evidence suggesting that low LDL levels per se do not increase the risk of death from cancer or non-cardiovascular causes.
Supplementary file 1
IL-6 promotes LDL-C catabolism by upregulating synthesis of LDL receptors
The evident alternative possibility to low LDL-C being pathogenic is that low or decreasing LDL-C may simply be serving as a marker for a factor or factors that is the true cause of excess non-cardiovascular mortality. It has long been known that LDL-C in humans and primates often declines acutely in the context of inflammation or trauma involving an acute-phase response—in the days following a myocardial infarction, for example.27–30 Indeed, studies with hepatocyte cultures show that interleukin (IL)-1β, tumour necrosis factor-α (TNFα), IL-6 and other cytokines elevated during inflammation can increase hepatocyte expression of the LDL receptor.31 32 Whereas IL-1β and TNFα do so by a mechanism involving modulation of intracellular cholesterol content, the effect of IL-6 in this regard is not dependent on cholesterol modulation.32 Nearly two decades ago, researchers reported that IL-6 increases the transcription of the LDL receptor gene, without altering the half-life of the transcript or of the resulting protein.32 This effect did not require new protein synthesis and was associated with increased binding of the transcription factors sterol regulatory element-binding protein (SREBP)1a/SREBP2 and Sp1 to their respective response elements in the promoter of the LDL receptor gene; hence, IL-6 likely is either increasing intranuclear transport of these transcription factors, or altering their configuration post-translationally in a way that enhances their DNA binding.
That this effect is of physiological importance is demonstrated by clinical studies showing that administration of a monoclonal antibody targeting and abolishing the bioactivity of the IL-6 receptor, tocilizumab, leads to an increase in plasma LDL-C of about 15%–20% in patients with rheumatoid arthritis.33–35 This effect has been traced to a decrease in fractional catabolic rate for LDL particles that parallels the increases in LDL level, pointing to a decrease in LDL catabolism as the chief basis for the observed increase in LDL-C; moreover, tocilizumab was found to decrease the expression of LDL receptors on hepatocytes in vitro.34 35 In contrast, a controlled study enrolling patients who had experienced a previous myocardial infarct and who had elevated C-reactive protein (CRP) found that treatment with the IL-1β-antagonist monoclonal antibody canakinumab did not influence LDL-C levels.36 And controlled studies in which patients with rheumatoid arthritis or ankylosing spondylitis were treated with the TNFα-antagonist monoclonal infliximab have observed at most a modest or transitory impact on LDL-C.37–39 Hence, these findings suggest that IL-6 is likely to be the chief driver of the LDL-C reductions seen during acute phase responses.
IL-6 correlates with increased total mortality in the elderly
These findings have been complemented by reports that plasma IL-6 levels correlate positively with global mortality in the elderly. Very recently, a meta-analysis of nine prospective studies enrolling over 9000 elderly subjects found that, when comparing the upper and lower categories of IL-6 in these studies, relative risk for total mortality was 1.49 (95% CI 1.33 to 1.67) and for cardiovascular mortality was 1.69 (95% CI 127 to 2.25) in those with higher IL-6.40 These findings were robust, holding up after adjustment for multiple covariants, and valid regardless of region or duration of follow-up. Although this study did not examine the association of IL-6 with non-cardiovascular mortality per se, the fact that non-cardiovascular death was responsible for about two-thirds of overall mortality suggests that risk for non-cardiovascular mortality was about 40% higher among those in the upper category of IL-6 when compared with that in the lower category.
The thesis that the association between low LDL cholesterol and increased cancer risk is mediated by IL-6 is difficult to square with prospective epidemiology that often has failed to correlate plasma IL-6 with subsequent cancer risk; most though not all41 42 meta-analyses examining this association in specific cancers have failed to confirm such a correlation.43–47 However, a recent meta-analysis confirmed that increased CRP predicts increased overall cancer risk.48 Most circulating CRP derives from the liver, and IL-6 is the primary stimulator of increased hepatic CRP production during an acute-phase response (although IL-1β amplifies this response); marked decreases of CRP are seen during tocilizumab therapy.49–51 Hence, CRP might be viewed as a marker for IL-6 bioactivity in the liver, which is what drives down LDL levels. Perhaps single measurements of IL-6 provide a rather imprecise assessment of IL-6 signalling in the liver.
Hence, it seems reasonable to postulate that, when low LDL-C points to increased non-cardiovascular mortality, it is simply serving as a marker for incipient pathologies, associated with inflammatory elevation of IL-6, which are driving the increase in mortality risk (see figure 1). If this is the case, then, in multiple-regression analyses which correct for plasma IL-6 levels, the association between LDL-C and non-cardiovascular mortality will be substantially attenuated, if not eliminated. A small residual correlation might nonetheless remain, reflecting a role for other factors—such as a premorbid or depression-linked decline in dietary intake, LDL clearance by an incipient cancer or liver dysfunction—capable of lowering LDL-C.
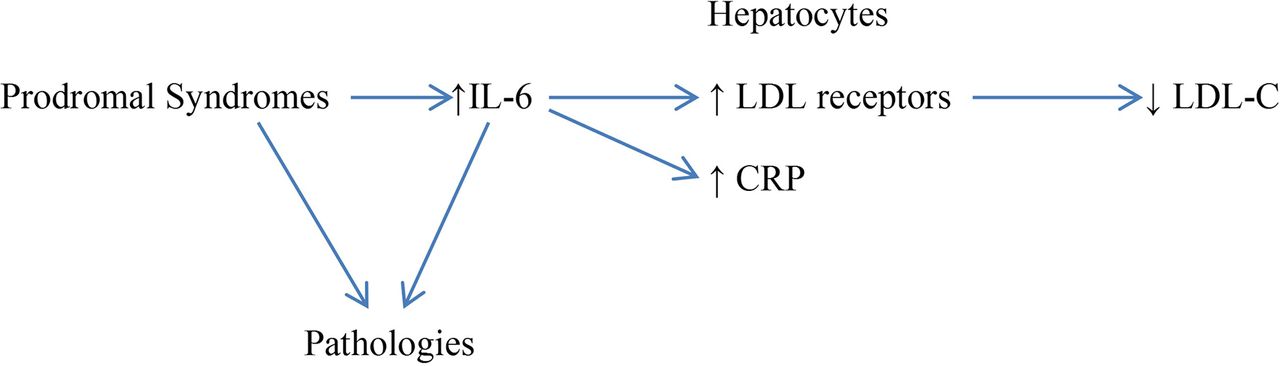
{kind=link}
Prodromal syndromes decrease LDL-C via increased IL-6.
IL-6 may also account for the loss of the positive association between LDL-C and cardiovascular risk noted in elderly populations. IL-6 levels tend to rise during the ageing process, even in the absence of evident infection or trauma; the greater incidence of abdominal obesity in the elderly, associated with macrophage activation in visceral fat, is likely partially responsible for this observation.52–55 Ershler has described IL-6 as ‘a cytokine for gerontologists’.52 As noted, higher IL-6 levels are predictive of increased cardiovascular risk in the elderly; this may reflect a mediating role for IL-6 in cardiovascular disease, but IL-6 may also serve as a marker for inflammation in visceral fat and in atherosclerotic lesions.56 57 The ability of elevated IL-6 to drive down LDL-C in the elderly should therefore tend to mask the continuing pathogenic role of LDL-C in atherogenesis and vascular events.
Conclusion
Whereas there is good reason to suspect that low LDL levels may increase risk for haemorrhagic stroke in hypertensives—while providing substantial protection from atherothrombotic events—relatively low LDL-C in the elderly has also been linked epidemiologically to increased risk for cancer and for non-cardiovascular mortality. However, this association is not likely to be causative, for reasons summarised in the online Supplementary Table 1. A decline in LDL-C is typically seen during an acute-phase response associated with trauma or infection. One of the chief mediators of the acute-phase response is IL-6. IL-6 increases the transcription of LDL receptors in cultured hepatocytes; conversely, treatment of patients with rheumatoid arthritis with the IL-6-antagonist monoclonal antibody tocilizumab has been found to raise their LDL-C by slowing LDL catabolism. Since monoclonal antagonists of IL-1β or of TNFα do not notably influence LDL-C, it is reasonable to conclude that IL-6 mediates the reduction of LDL-C observed during an acute-phase response. Importantly, meta-analysis has now confirmed that increased plasma IL-6 in the elderly is associated with a notable increase in all-cause and cardiovascular mortality; an increase in non-cardiovascular mortality can also be inferred from the data. Hence, it is proposed that when low LDL-C in the elderly predicts increased non-cardiovascular mortality, this low LDL-C is serving as a marker for inflammation-linked incipient pathologies, entailing increased production of IL-6, which are responsible for this increased mortality. This hypothesis can be tested in epidemiological studies that examine the association between LDL-C and mortality after statistical adjustment for IL-6 levels. It is further proposed that the age-related increase in IL-6 may explain, at least in part, the failure of LDL-C to correlate strongly with cardiovascular risk in the elderly—even though, as statin trials in the elderly demonstrate, LDL-C continues to promote vascular disease in this group.
The authors used keyword searches on Pubmed to locate the data discussed in this essay.
References
Footnotes
Contributors Both authors contributed to the final manuscript.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests JJD is author of The Salt Fix.
Patient consent Not required.
Provenance and peer review Not commissioned; externally peer reviewed.