Article Text

Abstract
Background Myocardial ischaemia–reperfusion injury is a major cause of mortality and morbidity in the developed world. Many approaches have been investigated to counteract the pathological consequences associated with acute myocardial infarction (AMI) and cardiac remodelling. It is accepted that inflammation, and therefore activation of the complement pathway, is a crucial step in the pathogenesis of this injury, and many attempts have been made to ameliorate the infarction and consequent dysfunction using anticomplement therapy, with mixed success. Recently, the lectin complement activation pathway involving the mannose-binding lectin-associated serine protease 2 (MASP-2) has been shown to be an important mediator of the inflammatory response in ischaemia/reperfusion injury in the heart. In this study, therefore, we aimed to investigate the feasibility of using monoclonal antibodies raised against MASP-2 in a murine model of AMI.
Methods Mice were injected with anti-MASP-2 antibody or control 18 hours prior to experimental infarction by ligation of the left anterior descending coronary artery for 30 min followed by 120 min reperfusion. The developed infarct was measured, and blood was collected for analysis of lectin pathway functional activity.
Results and conclusions We found that mice treated with anti-MASP-2 antibody had smaller infarcts than those treated with control antibody. We believe this may represent a valuable step forward in the protection of the myocardium against ischaemia–reperfusion injury.
- mouse
- myocardial ischaemia and infarction (ihd)
- inflammation
- haemodynamics
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Key questions
What is already known about this subject?
Mannose-binding lectin-associated serine protease 2 (MASP-2) has been shown to be an important mediator of the inflammatory response following acute myocardial infarction (AMI). Previous studies using a MASP-2-deficient mouse line showed that, in the context of acute injury, a significant reduction in post-AMI injury was afforded.
What does this study add?
Since global knockout of a gene is not practical in a clinical setting, we engineered a anti-MASP-2 antibody. We demonstrate that this antibody inhibits MASP-2 activity and, thus, create an environment at the time of myocardial ischaemia and reperfusion where MASP-2 activity is low, thus affording cardiac protection.
How might this impact on clinical practice?
While the present study focuses on the role of MASP-2 pathway on acute myocardial protection following an ischaemic insult (experimental AMI), we would anticipate that this protective effect would further affect postinfarction remodelling in a clinical setting. Future studies will address the long-term effect of MASP-2 inhibition on cardiac trophy in a more clinically relevant model of post myocardial infarction cardiac remodelling.
Introduction
Ischaemic heart disease is the leading cause of mortality in the developed world and contributes a significant burden of morbidity.1 2 Acute myocardial infarction (AMI), caused by inadequate blood flow through coronary arteries, leads to necrosis and can initiate chronic cardiac dysfunction and pathological cardiac remodelling.3 It is widely accepted that the mortality from AMI is directly related to infarct size and inversely related to the amount of myocardial salvage achieved following reperfusion.4 5 Likewise, detrimental myocardial remodelling, contractile dysfunction and electrophysiological instability are all greatly influenced by the volume of infarction.3 6–9 Although reperfusion following coronary artery occlusion is key to myocardial salvage, injury that continues following reperfusion, known as ‘reperfusion injury’, can be additive.10–12 The critical observations confirming the presence of reperfusion injury are the now innumerable interventions that limit ultimate infarction when given at the moment of reperfusion,13 since such interventions, by definition, are not present during ischaemia. Interventions that work at the moment on reperfusion include induced-hypothermia,14 altered reperfusate composition,15 16 staged, ramped or progressive reperfusion and,17 perhaps most convincing of all, anti-inflammatory agents that limit complement activation.18–20
It is now generally accepted that complement activation plays a key role in modulating myocardial ischaemia/reperfusion injury (MIRI) in animals and patients,19 21 in keeping with its deposition in areas of infarction in the human heart.22 23 Complement is a fundamental component of the immune system, providing protection against invading microorganisms through both antibody-dependent and antibody-independent mechanisms and mediates many cellular and humoural interactions within the immune response, including chemotaxis, phagocytosis, cell adhesion and B cell differentiation.24 Three different pathways initiate the complement cascade: the classical pathway (CP), the alternative pathway (AP) and the lectin pathway (LP). The LP is initiated by the binding of multimolecular activation complex to carbohydrate structures present on pathogens or glycocalyx pattern on mammalian cells. These complexes are composed of multimeric carbohydrate recognition subcomponents and the mannose-binding lectin-associated serine proteases 1, 2 and 3 (MASP-1, MASP-2 and MASP-3, respectively).25 26 This is a rapid first-line response towards pathogen-associated molecular patterns or altered-self molecular motifs known as damage-associated molecular patterns that are created during ischaemia/reperfusion injury.27–29 Analyses of the sera and plasma of MASP-2-deficient mice demonstrate that MASP-2 is the only enzyme required to activate complement via the LP, and neither MASP-1 nor MASP-3 are able to maintain LP activity in absence of MASP-2.30 We have previously demonstrated the importance of the MASP-2 in MIRI using these knockout mice, where deficiency significantly reduces MIRI and tissue loss in an experimental model of myocardial infarction, whereas C4-deficient mice were not protected from MIRI.31 These results indicate that long-term modulation of LP functional activity via therapeutic MASP-2 inhibition indicates a potential opportunity in reducing morbidity (and mortality) in MIRI. However, for this opportunity to be realised, the effect needs to be recapitulated by a therapeutic agent that transiently modulates the pathway. Such knowledge is a prerequisite in the clinical translation of anti-MASP-2 concept.
The aim, therefore, of the current study was to determine whether selective anti-MASP-2 monoclonal antibody therapy, at a dose optimised to systemically ablate the LP in mice, will reduce infarct sizes in an established model of in vivo MIRI. We adopted to pretreat experimental mice with inhibitory antibody therapy to ensure adequate effect of the dosing in accordance with some preliminary studies (data not shown) in order to assessMASP-2 inhibition on AMI.
Materials
Unless otherwise stated, all reagents, salts and stocks were from Sigma-Aldrich (Poole, UK). The 8–0 nylon sutures for ligation of the coronary artery were from Ethilon (Johnson & Johnson, Maidenhead, UK). Anti-MASP-2 monoclonal antibody (AbD04211) was isolated from Human Combinatorial Antibody Libraries (Serotec, AbD) using recombinant rat MASP-2A as antigens, as previously described.31–33 To optimise the cDNA of the VH and VL regions for mammalian gene expression, the genes were synthesised, and the variable domain fragments were subcloned into the pMORPH2_h/m_Ig vector series for human/mouse IgG2a. These vectors carry the mouse gamma 2a constant region or the mouse lambda constant region, respectively. Eukaryotic HKB11 cells were transfected with the IgG heavy and lambda light chain expression constructs by lipofection.34
Culture supernatants were harvested 3 days after transfection and clarified by centrifugation. Antibodies were purified from culture supernatants by single-step protein A affinity chromatography, followed by buffer exchange and sterile filtration using proprietary reagents (Serotec, AbD). Antibody concentration was determined by spectrophotometry (OD280 using a calculated extinction coefficient of 13.7). The final purity of antibody preparations was assessed by sodium dodecyl sulphate polyacrylamide elecrophoresis, and preparations were tested for endotoxin levels prior to administration into mice.
Methods
All animal experiments were carried out in accordance with Home Office regulations as detailed in the Home Office Guidance on the Operation of Animals (Scientific Procedures) Act 1986. HMSO (London), which mirrors those found in the Guide for the Care and Use of Laboratory Animals (NIH Publication No. 85–23, revised 1996). All experiments were carried out in a double-blinded manner using block randomisation.
MASP-2 inhibition using anti-MASP-2 monoclonal antibody
Dosing studies were carried out prior to carrying out the infarction arm of this study. Mice were administered 0.06–2.00 mg/kg AbD04211 or vehicle, and terminal blood samples were taken and used to determine LP-induced C4d deposition as an indication of the extent of MASP-2 inhibition as described below. From these initial experiments, a dosing regimen was established for the infarction study (see below).
Infarction study
Male C57BL/6 mice (n=46, average weight 31.8 g) were dosed with 1.0 mg/kg anti-MASP-2 mIgG2a (AbD04211), isotype matched control antibody (Anti-hen lysozyme mouse IgG2a with human variable domains, #MOR3207, AbD Serotec, Germany) or vehicle Phosphate-buffered saline (PBS) intraperitoneal (i.p.) 12–18 hrs prior to surgery according to a block randomisation process with a block size of 6. All mice were subjected to 30 min of coronary artery occlusion followed by 120 min of reperfusion, and infarct sizes were evaluated. Mice were anaesthetised with ketamine/metatomidine (100 mg/kg and 0.2 mg/kg, respectively), intubated and ventilated with 100% oxygen (Minivent II, Hugo Sachs, Germany). A left midthoracotomy was performed; the pericardium was opened, and a pericardial cradle was formed to move the heart slightly anteriorly. The left anterior descending (LAD) coronary artery was located, and a 8–0 monofilament suture with a round needle was then passed under the LAD. Coronary artery occlusion was achieved using the hanging weight system as previously described.35 Each end of the monofilament ligature was passed through a 2 mm long piece of a polythene PE-10 tube and attached to a mass of 1 g over two horizontally mounted movable metal rods. By elevation of the rods, the masses are suspended, and the occlusion of the LAD is instituted with a defined and constant pressure. LAD occlusion was verified by paleness of the area at risk. Reperfusion is achieved by lowering the rods until the tension of the ligature is relieved and reperfusion was verified by the same three criteria used to verify occlusion. Mice were excluded from further analysis if all three criteria were not met. At the end of reperfusion, citrate-treated (10%, v/v, 110 mM citrate, pH 8.3) blood samples were collected by direct cardiac puncture and plasma obtained by centrifugation (1300 × g for 15 min at 4°C) and retained for pharmacodynamic studies at −70°C.
Finally, the LAD was reoccluded, and 300 µL Evans Blue (2% w/v in 0.9% v/w NaCl) was injected into the jugular vein to delineate the area at risk. Hearts were rapidly excised, mounted in agarose and sliced in an ice bath (4°C) into 750 µm sections from apex to base. All slices were incubated at 37° C for 20 min with 3% w/v 2,3,5-triphenyltetrazolium chloride (Sigma-Aldrich) dissolved in 0.1 M Na2HPO4/NaH2PO4 buffer adjusted to pH 7.4. Slices were fixed overnight in 10% formaldehyde, and then placed between two cover slips and digitally imaged using a high-resolution optical scanner (Epson, UK). Images were analysed using Sigma-Scan V.5.0 software, and the size of infarcted area (pale), LV area at risk (red) and normally perfused LV zone (blue) was outlined in each section by identification of their appearance and borders. Areas were quantified on both sides of each slice and averaged. Infarct size was calculated as a percentage of risk zone (RZ) for each heart. Any mouse that did not survive the surgical protocol or have a measureable RZ by Evans Blue staining was excluded from further analysis. One-way analysis of variance for multiple comparisons was used to test for differences between treatment groups followed by a Bonferroni post hoc test for pair wise comparisons. Significant differences were assumed when P<0.05.
Complement assay
LP inhibitory activity of anti-MASP-2 mAb was assessed in a C4 deposition assay using dilute mouse serum. Appropriate dilutions of anti-MASP-2 antibody were mixed with 0.5% normal mouse plasma in ‘CaMg-GVB’ buffer (gelatine veronal buffer containing 2.0 mM CaCl2 and 1.0 mM MgCl2) supplemented with 1.0 mg/mL of human C4 protein. One microgram of mannan in coating buffer (15 mM Na2CO3, 35 mM NaHCO3, pH 9.6) was added to each well of an ELISA plate (Nunc MaxiSorb, Nunc International, Rochester, New York, USA) and incubated overnight at 4°C. After a 1 hour preincubation on ice, mixtures are transferred into mannan-coated ELISA plates, and complement activation is initiated by transferring plates to a 37°C water bath. Following 1 hour incubation, reactions are stopped in an ice/water bath, followed by three washes with cold PBS Tween-20 (0.05% Tween-20 in PBS) and two washes in PBS. To detect C4b deposition, wells are incubated with biotinylated anti-C4 antibody (diluted 1:3000 in PBS containing 2.0 mg/mL bovine serum albumin, BSA) for 1 hour at room temperature (RT). After washing five times with PBS, wells are incubated with 0.1 µg/mL of peroxidase-conjugated streptavidin (Pierce, diluted in PBS containing 2.0 mg/mL BSA) for 1 hour at RT, washed with PBS and developed with TMB substrate and measured as absorbance at 450 nm after addition of 20 µL 100 mM sulphuric acid to each well using a microplate absorbance spectrophotometer (Labsystems Multiscan Plus, Fischer Scientific, Pittsburgh, USA).
To assess the extent of LP inhibition resulting from anti-MASP-2 or isotype control antibody administration, an ex vivo LP assay using minimally diluted mouse serum (90% final serum concentration) was employed. To prepare serum, mouse citrate plasma samples collected at various time points after test article administration were supplemented 10 mM with CaCl2 and incubated for 1 hour at 37°C to allow fibrin clot formation. The samples were then frozen at −70°C, thawed and centrifuged 14 000 rpm for five min at 5°C in the refrigerated table-top centrifuge to remove the fibrin clot. Clear serum samples were then transferred to a polypropylene plate on ice. Serum from three control animals was pooled to generate reference LP activity values. To one aliquot of this serum pool anti-MASP-2 blocking mAb was added (50 nM final concentration), while the other aliquot was supplemented with an equal volume of PBS. These samples were used to generate the 0% and 100% serum LP activity values, respectively. LP activation was initiated by transfer of 50 µL of mouse serum onto ELISA plates coated with mannan followed by 90 min incubation at 0°C in an ice/water bath. To stop reactions samples were aspirated and ice-cold EDTA–Tween–PBS buffer (20 mM EDTA, 0.5% Tween 20 in PBS) was added to wells. Wells were washed three times with ice-cold EDTA–Tween–PBS buffer followed by two washes with PBS at RT. To detect C4b deposition, wells were processed as described above.
Results and conclusions
Out of 46 mice subject to experimental infarction, 41 completed the study and were evaluated. Five mice were excluded from analysis due to premature death (n=4) or because the inclusion criteria were not met (n=1). The exclusions were from all three groups: the anti-MASP-2 (n=2), isotype antibody (n=1) and vehicle (n=2) group. The outcome of in vitro assays evaluating LP inhibition following administration of the anti-MASP-2 mAb (AbD04211) antibody is shown in figure 1A–C. We found that a dose of 1.0 mg/kg i.p. was effective it inhibiting LP-dependent C4 deposition below the detection threshold of the assay at the first test point (6 hours) and was sustained for up to one week post-treatment (figure 1C). The data from the infarction study are presented in figure 2. There was no difference in the RZ as a percentage of total left ventricle volume (%LV) between groups (P=0.8), indicating comparable magnitude of myocardial ischaemia (figure 1A). Infarcts, however, were significantly smaller in the AbD04211-treated group (15%±1.0%) compared with control isotype (26.1%±1.9%) and vehicle (22.4%±1.8%) treated groups (P<0.005 and P<0.01, respectively (figure 2B)). A comparison of infarct sizes expressed as percentage of the RZ, unsurprisingly, yielded similar results (figure 2C,D). Infarcts were significantly smaller in animals treated with AbD04211 (23.1%±1.3%) compared with isotype control (38.5%±2.1%) or vehicle (34.3%±2%), and this effect was evident across a range of RZs (figure 2D).
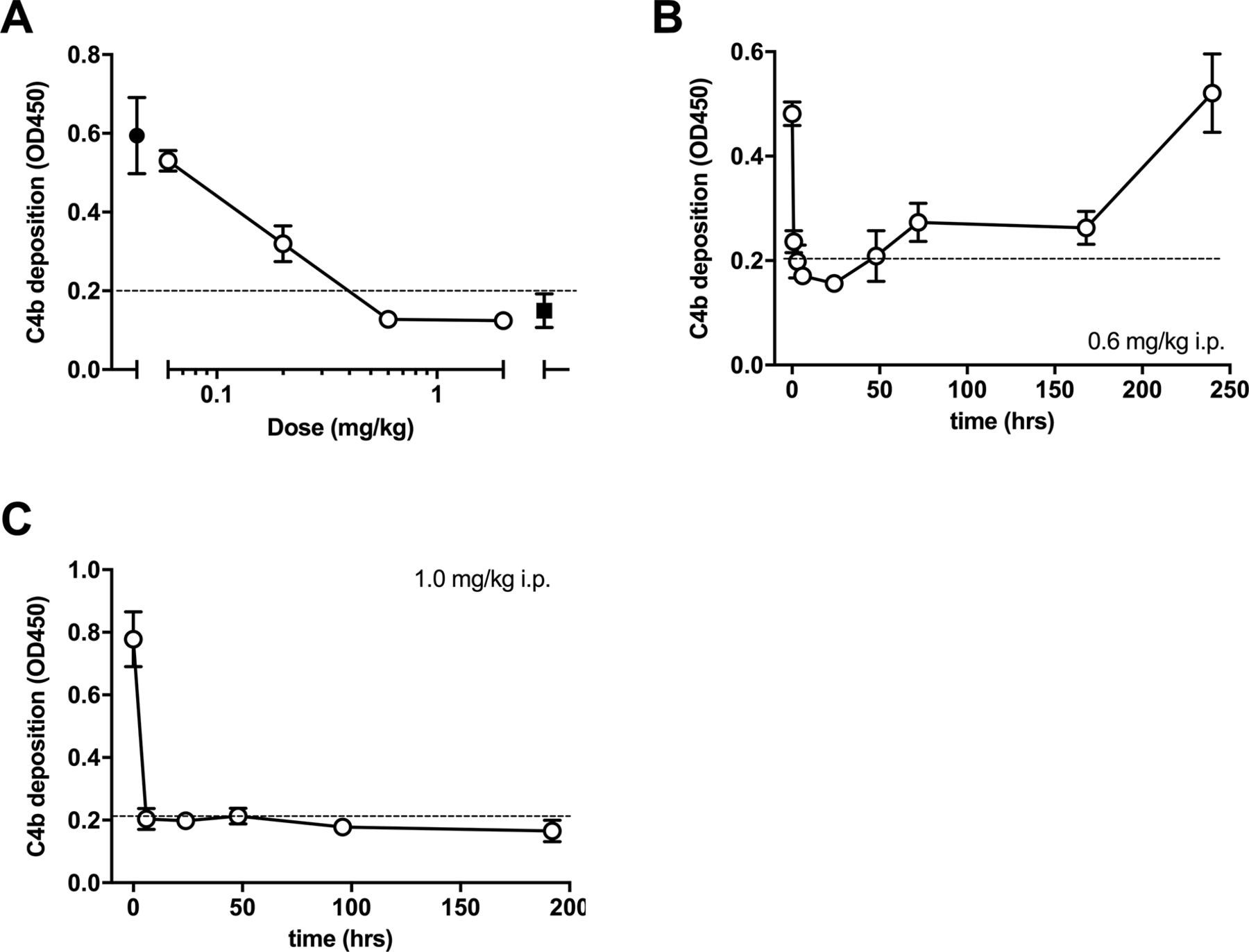
Dose–response of lectin pathway (LP) inhibition following administration of anti-MASP-2 mAb (AbD04211) into C27BL/6 mice: (A) AbD04211 (0.06, 0.2, 0.6 or 2.0 mg/kg) or vehicle (PBS) was injected (i.p.) into mice and terminal plasma samples collected 6 hours after dosing. LP was assessed in 90% serum using C4b deposition onto mannan-coated plates as readout. Each data point represents mean±SD of three mice. C4 deposition in serum from mice dosed with vehicle (black-filled circle) and C4 deposition using the same serum supplemented with 50 nM of AbB04211 in vitro (black-filled square) are shown for reference. Time-course of LP ablation and recovery following administration (i.p.) of 0.6 mg/kg (B) or 1 mg/kg (C) AbD04211 into mice. LP was assessed in 90% mouse serum using C4b deposition onto mannan-coated plates as readout. Each data point represents mean±SD of three mice. Dashed lined shows baseline C4 deposition independent of LP (naive mouse serum supplemented with 50 nM of AbD04211 in vitro). MASP-2, mannose-binding lectin-associated serine protease 2.

{kind=link}
{kind=link}
Infarct and risk zone (RZ) assessments. Mice were subjected to 30 min ischaemia followed by 120 min reperfusion in vivo. RZ, infarct size (INF) and left ventricle (LV) size were measured by Evans blue and tetrazolium staining. (A) RZs determined in mice pretreated with vehicle (PBS), isotype control Ab or AbB04211; (B) INF as a percentage of LV mass and (C) INF as a percentage of RZ in the three groups. (D) INF versus RZ in all the experimental mice. Data are presented as total area at risk and INF as a percentage of LV volume. Results are mean±SD of 13–15 independent experiments. **P<0.01; ***P<0.005 as indicated.
Injury resulting from myocardial ischaemia leads to a proinflammatory response that will undoubtedly involve activation of the three complement activation pathways. Indeed, a recent clinical study has demonstrated that MASP-2 activation is involved in ischaemia-related necrotic myocardial injury in human subjects following MI.36 Our data presented here demonstrate that treatment with an anti-MASP-2 antibody blocking the LP of complement activation prior to ischaemia reduces the resultant infarction and increases myocardial salvage. One may suppose that anti-MASP-2 therapy may also be useful to reduce infarct size, preserve contractile function and improve long-term outcomes in patients with myocardial infarction undergoing percutaneous coronary intervention following occlusion of their coronary artery, although this is outside of the scope of this study, it will be the focus of future studies in a more clinically relevant model of post-AMI remodelling with concurrent treatment with MASP-2 inhibition therapy.
In conclusion, we have shown that the MASP-2 dependent activation of complement critically contributes to continued myocardial injury during reperfusion following AMI. Ultimately, this injury would manifest in adverse postinfarction remodelling and poor contractile performance, clinical correlates of late onset cardiac failure.7 9 We hypothesise that the therapeutic application of MASP-2 inhibition provides a significant window of opportunity to reduce the morbidity and mortality associated with MI.
References
Footnotes
Contributors JEC was responsible for the experimental design, laboratory work and undertaking of the mouse experiments. JEC also wrote and edited the manuscript and prepared the figures. MSM carried out review of the manuscript. TD provided the Ab for the study as well as reviewing data and analysis and the manuscript. WS reviewed the manuscript and provided support in the conduct of MASP-2 experiments.
Competing interests JC and MM have no interest to declare. TD is an employee and stockholder of Omeros Corp. WS is a consultant and receives research support from Omeros Corp. This study was funded partly by Omeros Corp.
Provenance and peer review Not commissioned; internally peer reviewed.