Article Text

Abstract
Heart failure has been recognised for years but the complete picture has been difficult to clearly understand. This article aims to try and put forward a proposed mechanistic explanation to encompass all that we see within the clinical heart failure syndrome using supporting published evidence. The aim of the article is to link, using published evidence, all the known varieties of heart failure into a spectrum that is explained by simple interlinked processes. In addition, the concept of routinely looking for reversibility of left ventricular dysfunction is introduced.
- heart failure
- hfpef
- hfref
- damage
- fatigue
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Traditional understanding of heart failure
Historically, heart failure was thought to be a disorder of its pumping function and the ejection fraction (EF); therefore, these became an important component and determinant of the condition. The increasing prevalence of heart failure with preserved ejection fraction (HFPEF) and the current lack of disease-modifying therapy for this group of patients raise the question again of whether we truly understand the mechanisms that drive the entire spectrum of the heart failure syndrome. How can all the different described types of heart failure be linked up to create a spectrum and a story that is easily understandable? Can we link the relationship of the core mechanism of heart failure with EF and then with the types of HF such as heart failure with reduced ejection fraction (HFREF), HFPEF or even with the old classification of high-output, low-output, forward and backward failure, right and left heart failure? Unless we progress further in answering these questions, discovering treatment for the entire heart failure population will be difficult.
Damage, fatigue and injury concept
For chronic heart failure, there may be three basic but linked mechanisms that exist to cause the heart failure: damage, fatigue and injury.
The damage mechanism is very evident in the common myocardial infarctions1 or some toxic/inflammatory cardiomyopathies (eg, anthracycline chemotherapy-induced cardiomyopathy or chronic myocarditis)2 and is naturally associated with reduced EF (HFREF). Here, there is a large amount of irreversible myocardial damage leading subsequently to fibrosis; the heart failure is systolic and is due to pump failure.
However, myocardial injury can often be transient, acute and limited, such as with acute viral myocarditis causing myocardial inflammation and oedema, or other reversible cardiomyopathies (injury of an undefined nature in peripartum cardiomyopathy, a sympathoexcitation-induced injury in Takotsubo cardiomyopathy or a hypermetabolic injury in heart failure related to a thyroid storm).3 It would not be unreasonable to assume that the reversible forms of left ventricular (LV) systolic dysfunction are the ones where injury has played a part but significant damage has not occurred (the injury group). There could be variable amounts of injury and damage caused by alcohol, chemotherapeutic agents or other drugs, catecholamines, infections and so on with the final outcome on LV function determined by the proportion of injury versus damage. Genetic causes or myocardial infiltration would most likely also fall under the injury versus damage headings.
Myocardial fatigue may be the cause of heart failure if the ventricle is having to pump against high vascular resistance.4 5 This may be the main mechanism of HF in patients with HFPEF as there is increasing evidence that microvascular dysfunction6 7 is probably the key operating mechanism in this variety of heart failure. Data have been presented to suggest that patients with HFPEF are likely to be progressing from a stage of preclinical diastolic dysfunction to the HFPEF stage with increasing left ventricular end-diastolic pressure (LVEDP) as assessed by echocardiographic E/E′. 8 It can be speculated that the elevated LVEDP is related to the LV struggling to pump into a high-pressure vascular circuit that is a result of microvascular dysfunction. Ageing9–11 has an important role in gradually increasing arterial resistance as have comorbidities12 such as hypertension13, diabetes14 15 and chronic kidney disease.16 17 There is probably additional contribution to this vascular ageing from changes in the heart itself of both microvascular rarefaction and myocardial fibrosis (including perivascular fibrosis), which reduces coronary flow reserve resulting in LV diastolic dysfunction.18
Cardiac fatigue has been described in athletes,19 20 is reversible and appears to affect ventricular diastolic function21 more than systolic function. The above hypothesis is, therefore, plausible. While fatigue in the early stages would manifest as increased LVEDP with preserved LV systolic function, prolonged or severe fatigue would naturally lead to systolic dysfunction and pump failure (which may be reversible if the fatigue can be tackled by say controlling poorly controlled blood pressure22) probably because of the development of focal and diffuse myocardial fibrosis that accompanies severe fatigue. Systemic hypertension, the the most common and most important cause of HFPEF, causes myocardial fatigue as a result of the enhanced vascular stiffness against which the LV has to pump. Another parallel example of fatigue would be HF related to conditions such as hypertrophic obstructive cardiomyopathy (HOCM)23 and severe aortic stenosis,24 which increase LVEDP by causing intramyocardial resistance to blood flow at the level of the left ventricular outflow tract and the stenotic aortic valve, respectively. In these conditions, the heart failure is usually of the HFPEF type as the ventricle is having to pump against high resistance (which is at the cardiac rather than at the vascular level), with a consequent increase in LVEDP. Interestingly, intervention to reduce the high resistance, such as an aortic valve replacement for severe aortic stenosis, can result in recovery of the LV fatigue,24 particularly in those without myocardial fibrosis,25 presumably as the myocardium in these patients is in a state of chronic fatigue that has not reached an advanced stage that is accompanied by fibrosis. On the other hand, advanced and prolonged fatigue can result in irreversible myocardial changes of hypertrophy and fibrosis26 as seen in some patients with severe untackled aortic stenosis with no cardiac reserve. In other words, recovery is linked to the stage of fatigue. Advanced fatigue with extensive fibrosis is unlikely to recover.
Advanced myocardial fibrosis that is seen in both patients with aortic stenosis (AS)27 and patients with HOCM28 may be a consequence of chronic progressive fatigue through myocyte loss, but in HOCM, myocardial abnormalities including hypertrophy and fibrosis also have a genetic basis. In non-obstructive hypertrophic cardiomyopathy, the hypertrophy and fibrosis are genetically determined and lead to a purely cardiac cause of HFPEF due to LV diastolic dysfunction with little contribution from vascular stiffness.
Looking at the phenotypes of AS from a fatigue angle, AS with preserved LVEF would correspond to early LV fatigue with preserved EF and stroke volume at the expense of elevated LVEDP. Later when LVEDP rises substantially, LV filling becomes impaired leading to low-flow, low-gradient AS with preserved LVEF, but the stroke volume and cardiac output would have reduced. Finally, as advanced fatigue sets in, there is fibrosis due to myocyte loss, LVEF starts to fall and the ventricle starts to dilate leading ultimately to a dilated ventricle with reduced EF, the so-called low-flow, low-gradient severe AS with depressed EF. A ventricle suffering from uncorrected advanced fatigue can, therefore, end up looking like one that is a result of damage. The pathophysiology of the end stage, however, is likely to be different in the fatigued ventricle with focal and diffuse fibrosis occurring in a chronic slow fashion as opposed to fairly rapid fibrosis that occurs due to damage during infarction or inflammation.
High-output cardiac failure29 would also be an example of fatigue-related heart failure as a result of the myocardium’s inability to cope with an enhanced volume of blood.
Where does heart failure with mid-range ejection fraction fit into this?
The European Society of Cardiology has introduced the new category of heart failure with mid-range ejection fraction (HFMREF) in its new classification in 201630 defined as signs and symptoms of heart failure with LVEF between 40% and 49%. It has been suggested that HFMREF is a unique phenotype of heart failure, and studies on this group have shown mortality and readmission rates between that seen in HFREF and HFPEF patients.31 Sticking with the same hypothesis that HFREF is mainly a myocardial problem that is damage related and HFPEF mainly a vascular problem that is fatigue related, HFMREF may be HFPEF that is gradually dropping LVEF due to prolonged or increasing fatigue. In other words, HFMREF may be simply another fatigue-related vascular issue but at a more advanced level than HFPEF. Of course, two other components of HFMREF would be patients with HFREF who have suffered mild damage or patients with HFREF who originally had severe damage but have now improved their EF. These subsets were assessed in a recent study32 in which the demographics of the HFMREF cohort were found to be heterogeneous, with more coronary artery disease in the HFMREF improved EF group (improved from below EF <40%) and more hypertension and diastolic dysfunction in the HFMREF deteriorated EF group (deteriorated from EF >50%). The other two groups compared were HFREF and HFPEF. HFMREF improved EF patients had significantly (p<0.001) better clinical outcomes relative to matched patients with HF and reduced ejection fraction, and significantly (p<0.01) improved clinical outcomes relative to HFMREF deteriorated patients, whereas clinical outcomes of the HFMREF deteriorated subgroup of patients were not significantly different from matched patients with HF with preserved ejection fraction. These results appear to suggest that the patients with HFMREF that are HFREF showing improvement of EF do better than patients with HFPEF with reducing EF.
Assessment of arterial stiffness
Of a number of non-invasive methods that are available to assess vascular stiffness, the two commonly used ones are pulse wave velocity (PWV) and augmentation index (AI).33 The elasticity of a segmental artery (say carotid–femoral) is assessed by PWV. A pulse wave is generated by each cardiac contraction, which travels distally and can be measured by the distance travelled by the pulse wave divided by the time taken between two regions such as carotid to femoral. More the arterial stiffness, more the PWV. Carotid–femoral PWV measures central arterial stiffness,34 whereas brachial-ankle PWV measures both central and peripheral arterial stiffness.35 As changes in blood pressure (BP) can affect these measurements, a new cardio-ankle vascular index (CAVI)36 has been used that is independent of BP. CAVI uses measured PWV adjusted by a stiffness index β. Particularly in the elderly, arterial stiffness independently predicts cardiovascular morbidity and mortality.37–39
AI is derived from the interaction between the forward pulse wave and the reflected wave and is a measure of resistance of the entire arterial tree.40 It increases with increasing arterial stiffness41 and is influenced both by BP33 and heart rate.42 Although both PWV and AI increase with age, changes in AI are more prominent in younger people below <50 years of age and PWV changes are more marked in those older than 50 years43. In women after menopause, arterial resistance rises sharply and is higher than in men.44 45
A review of the literature on vascular stiffness does support the hypothesis that ageing-related arterial stiffness46 coupled with comorbidities such as hypertension47 and diabetes,48 which enhance the stiffness, can lead to HFPEF. Support of this also comes from a small non-invasive study comparing arterial compliance, venous capacitance and microvascular function in three patients’ groups comprising HFPEF, HFREF and control (non-HF). The study found that there was significantly reduced vascular compliance (increased arterial stiffness, decreased venous capacitance) in the HFPEF group compared with the HFREF and control groups (p<0.05). 49
How might mechanistic thinking in relation to investigations for HF help?
Thinking of damage, fatigue or injury while investigating someone with heart failure may be clinically useful to try and predict if the heart failure may be reversible in an individual and thereby link to prognosis in that individual. A clinician may identify the aetiological factors that might predict the degree of injury versus damage on a cardiac MRI scan and make a better judgement of the long-term outcome. A good example would be the degree of myocardial oedema versus patchy scarring seen on a cardiac MRI scan (CMR) in a patient with myocarditis, which helps to assess just this. A heavily scarred LV from a large myocardial infarct seen on CMR is quickly judged as having had a large amount of damage that would not be reversible. On the other hand, the finding of elevated LVEDP on echocardiography with preserved LVEF along with elevated brain natriuretic peptide but with no infarction or infiltration on cardiac MRI (and normal perfusion) could point the finger at fatigue-related HFPEF, which can be potentially reversed if a reversible precipitating factor (such as poorly controlled blood pressure or diabetes) can be identified or new research is able to find a cure for microvascular dysfunction. In the case of HFMREF, the finding of significant infarction could separate out the damage-related HFMREF (where HFREF drugs should still work) from the fatigue-related HFMREF (where HFREF drugs are unlikely to work). Mechanistic thinking would also help with understanding why the EF is preserved, low or mid range in a patient with heart failure. For example, a CMR showing reduced LVEF with no infarction but reduced perfusion would be able to explain the HFREF by a mechanism of fatigue (muscle weakness) but due to reduced perfusion to the cardiac muscle rather than fatigue secondary to elevated arterial resistance.
Assessing for reversibility of LV dysfunction
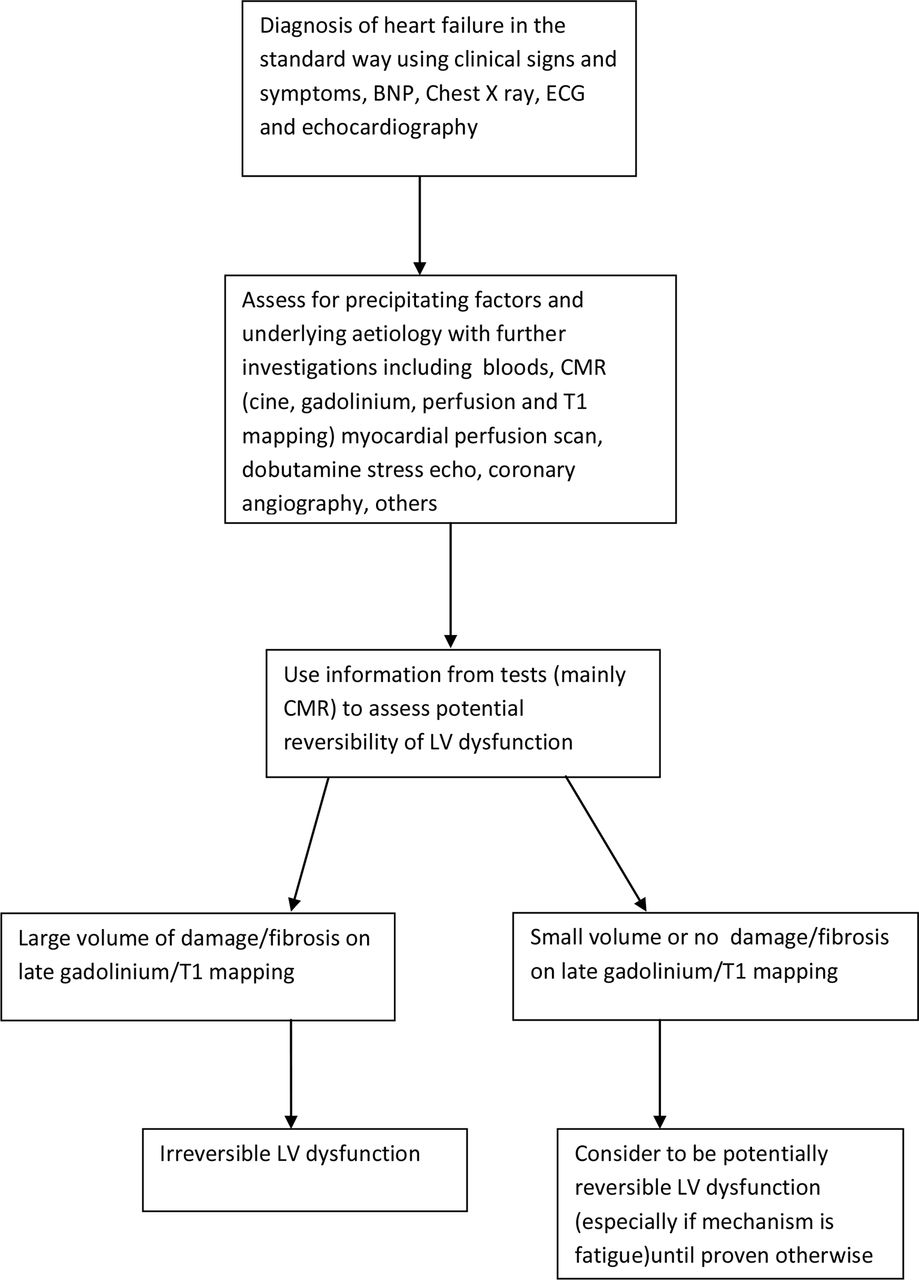
While assessing a patient with heart failure, an important aim of a heart failure physician is to identify whether the heart failure may be due to a reversible process as identification of reversible factors may lead to effective treatment of these and subsequent complete cure of the HF. Looking for underlying aetiologies and precipitating factors forms part of this assessment. The assessment could be further aided by linking in the mechanism of the heart failure (damage, fatigue or injury) with evidence gathered from available investigations such as a cardiac MRI scan, myocardial perfusion scan, dobutamine stress echo and so on. Focal replacement fibrosis, corresponding to areas of myocyte loss, is seen well by delayed gadolinium enhancement on CMR and linked with irreversible impairment of LV systolic function and a worse prognosis. 50 51 A novel method to detect a different type of fibrosis- diffuse interstitial myocardial fibrosis, using T1 mapping by CMR,52 53 has also now been developed and will aid this process of assessment for reversible LV dysfunction. While diffuse fibrosis by T1 mapping is also a marker of worse prognosis, it may not be as concerning as focal fibrosis and there is current interest in it as a therapeutic tool54. T1 mapping is likely to become a really well-established tool for cardiac disease assessment55 in the near future. The common reversible cardiomyopathies56 (tachycardia induced, peripartum, inflammatory/infectious, hyperthyroidism induced, Takotsubo and chronic illness induced such as with cirrhosis, obesity, uraemia) could all be mechanistically assessed in this manner to further enhance our expected prediction of the degree of reversibility of LV systolic function in individual cases. Knowledge on the degree of reversibility of LV dysfunction is likely to improve decision making on treatment options (for example, a large amount of expected reversibility should point towards intensive care treatment in the acute setting and consideration for mechanical circulatory support rather than palliative care in the appropriate patient). A proposed pathway of assessment of heart failure to discern reversibility is provided in figure 1 as a starting point in this exercise.

{kind=link}
Suggested plan of assessment of reversibility of LV dysfunction in a patient with heart failure. Although not perfect by any means, this is a starting point. BNP, brain natriuretic peptide; CMR, cardiac MRI scan; LV, left ventricle.
Conclusion
Systolic or diastolic dysfunction (or ejection fraction) may not be as important as the underlying mechanism of damage, fatigue or injury in the heart failure syndrome in understanding the core mechanism of heart failure in an individual patient. There will often be a mixture of systolic and diastolic dysfunctions in many patients with a predominance of diastolic impairment in the fatigue variety and of systolic dysfunction in the damage variety. As understanding of the entire heart failure syndrome improves, the classification may change to reduce its reliance on ejection fraction and may contain new elements such as arterial resistance, ventricular end-diastolic pressure and perhaps even basic mechanistic concepts such as damage, fatigue and injury. Further research focusing particularly on myocardial fatigue in patients with HFPEF will determine whether this mechanistic model of damage, fatigue and injury truly stands up to scrutiny.
References
Footnotes
Contributors The entire article is conceived and written by PB alone.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement No additional data available.