Article Text

Abstract
The life of a human being originates as a single cell which, under the influence of certain factors, divides sequentially into multiple cells that subsequently become committed to develop and differentiate into the different structures and organs. Alterations occurring early on in the development process may lead to fetal demise in utero. Conversely, abnormalities at later stages may result in structural and/or functional abnormalities of varying severities. The cardiovascular system and skin share certain developmental and structural factors; therefore, it is not surprising to find several inherited syndromes with both cardiac and skin manifestations. Here, we will review the overlapping pathways in the development of the skin and heart, as well as the resulting syndromes. We will also highlight several cutaneous clues that may help physicians screen and uncover cardiac anomalies that may be otherwise hidden and result in sudden cardiac death.
- Cardiocutaneous
- Dermatologic
- Congenital
- Syndromes
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Structure and function of desmosomes
The skin and heart—two seemingly unrelated organs—share a crucial characteristic: both are subjected to shear mechanical stress, and thus should be resilient as to accommodate this stress in a flexible way, in order to maintain tissue integrity. One of the common denominators between the two is the presence of desmosomes, which are found in the epidermis and the myocardium and are thought to play a major role in accommodating this stress. ‘Desmosomes’ is a composition of the Greek words ‘desmos’ meaning bond and ‘soma’ meaning body. Their main function is, as the name implies, to secure bonds between the cells and thus resist forces of stress. Both the heart and skin have ultrastructurally similar desmosomes made up mainly of the desmoplakin and plakoglobin proteins (figure 1).1
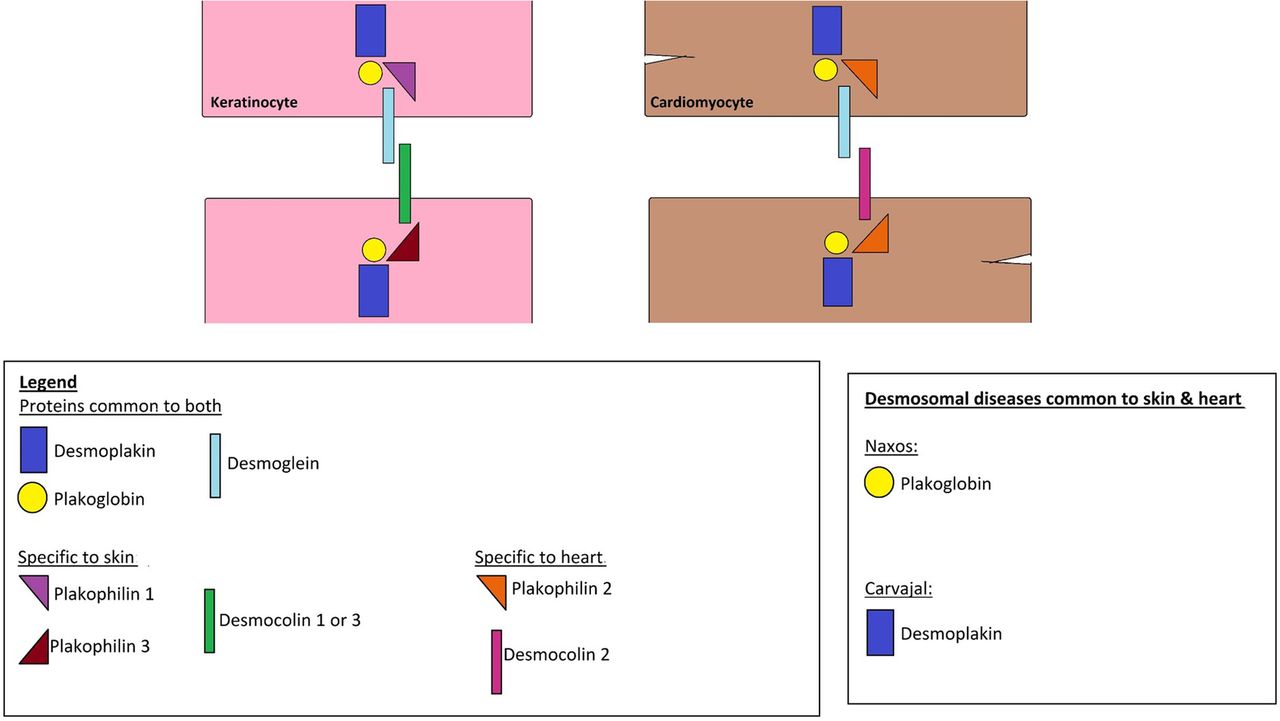
Schematic representation of structural proteins found in keratinocytes and cardiomyocytes.
Structurally, desmosomes are made up of adhesion proteins of the Cadherin family that span the cellular membranes of the cells linking them to the intracellular keratin filaments. The desmosomal proteins common to both the epidermis and myocardium are the desmocolins DSC2, DSG2 (low levels in the epidermis), plakophilin PKP2, desmoplakin DSP, plakoglobin PG and plectin. Nevertheless, the skin and heart also do have unique proteins for each: the desmosomal cadherins desmoglein DSG1, DSG3, DSG4, DSC1, DSC3 and the glycoprotein corneodesmosin (CDSN) form the extracellular linkage in the epidermis while in the myocardium, DSG2 and DSC2 function as the extracellular linkers. Furthermore, PKP1 and PKP3 are the major epidermal PKPs, whereas PKP2 is the only PKP present in cardiac tissue.1 ,2
It is worth noting that mutations in the same gene may lead to different syndromes because of several factors, such as the location of the mutation, the type of the mutation, whether it affects regulators, transcription factors or binding sites, as well as the individual's genetic blueprint that interacts with environmental factors resulting in unique or overlapping phenotypes.
Embryonic origin
Most cardiac cells originate from the mesoderm, while the skin tissue is derived from both the mesoderm (dermis) and the ectoderm (epidermis). Paradoxically, many inherited diseases have both cardiac and cutaneous phenotypes suggesting a potential common progenitor cell behind this observation. It was not until recently that a study conducted by Hwang et al3 identified a novel population of melanocyte-like cells in the hearts of mice and humans, using a Cre-inducible melanocyte-specific marker. These cardiac melanocytes express melanin-synthesis enzymes and morphologically resemble melanocytes of the skin. While sparsely distributed throughout the mice atrium where they comprise <0.1% of all atrial cells, cardiac melanocytes were found to be present in anatomic locations from which atrial arrhythmias often originate in humans. These include the pulmonary veins, atrioventricular annulus, posterior left atrium and foramen ovale.3
Another study conducted by Brito and Kos4 identified large numbers of melanoblasts in the atrioventricular endocardial cushions in the embryonic period that persisted into adulthood in the atrioventricular valves. Cardiac melanocytes were absent in the atrioventricular valves of KIT and EDNRB mutant mice, while large numbers were observed in transgenic mice that overexpress endothelin 3. KIT and EDNRB signalling are crucial players that affect the survival, development and migration of cutaneous melanocytes. These findings indicate that cardiac melanocytes depend on the same signalling pathways required by cutaneous melanocytes, and may even generate from the same precursor population.4 Although the exact mechanism underlying cardiocutaneous overlap manifestations has not been well defined until now, the findings stated above support the existence of many of the diseases (discussed below) that encompass cardiac as well as melanocytic manifestations.
Here, we will review the overlapping pathways in the development of the skin and heart, as well as the resulting syndromes. We will also highlight several cutaneous clues that may help physicians screen and uncover cardiac anomalies that may be otherwise hidden and result in sudden cardiac death.
Cardiocutaneous syndromes
Carney complex
Carney complex (CNC) is a rare syndrome characterised by multiple neoplasms of the heart (myxomas), skin and endocrine system, as well as lentigines, pigmented lesions of the skin and mucosae. More than half the cases are familial, inherited in an autosomal-dominant fashion, with almost 100% penetrance. The rest occur sporadically as a result of a de novo mutation.
CNC is a rare disease of unknown prevalence that typically affects females more than males. It is usually diagnosed starting in the 2nd year of life and as late as the 5th decade of life, with a median age of 20.5
It is important to recognise diagnostic pigmented cutaneous manifestations in CNC, as they can lead to the early detection of the disease and thus the prevention of life-threatening complications of CNC related to cardiac myxomas and endocrine abnormalities.
Cutaneous manifestations
The cutaneous manifestations are the most definitive suggestion of CNC, as more than 80% of patients are found to have pigmented spots or skin growths. These can be categorised into three major manifestations:
Lentigines: these are small flat macules, brown to black in colour, typically located on the face, lips, genital area and mucosa. They usually appear before puberty, but increase in number and pigment intensity during and after adolescence.6
Cutaneous or mucosal myxomas: 30–55% of patients with CNC are reported to have cutaneous myxomas, usually in the eyelid, external ear canal, nipples and genitalia. These present as asymptomatic, sessile, small, dark pink papules and large, finger-like, pedunculated lesions.5
Multiple blue nevi or epithelioid blue nevus: small (usually <5 mm), blue to black–coloured marks with a circular or star-shaped appearance. Although very rare in the general population, an epithelioid blue nevus is highly associated with CNC and should direct clinicians towards considering CNC as a diagnosis.7
Cafe’-au-lait spots, irregular depigmented areas, many compound and, rarely, Spitz nevi have also been reported.5
Cardiac manifestations
The most common non-cutaneous lesions in CNC are cardiac myxomas (20–40% of patients). As opposed to sporadic cardiac myxomas that develop almost exclusively in the left atrium and more frequently in women, those of CNC occur anywhere in the heart and equally in men and women. Cardiac myxomas are of significance as they are responsible for more than 50% of mortality from the disease, via occlusion of a valve, leading to sudden death. They may also present with strokes and/or heart failure, as a result of blood flow obstruction or systemic emboli.6 ,7
Other manifestations
Other frequently encountered manifestations of CNC include facial and palpebral lentigines, eyelid myxomas and pigmentation of the semilunar fold. Endocrinological findings include asymptomatic elevation of the growth hormone, insulin-like growth factor 1, or prolactin, primary pigmented nodular adrenocortical disease (which leads to overproduction of cortisol) and thyroid nodules (most often benign). Patients may also have psammomatous melanotic schwannomas (nerve sheath tumours in the central and peripheral nervous systems), testicular tumours, breast myxomas and ovarian tumours.6 ,7
Genetics
Most cases of CNC are attributed to mutations in CNC1, the PRKAR1A gene coding for the regulatory subunit type I α of the protein kinase A (PKA, cAMP-dependent protein kinase) enzyme. Studies have shown the involvement of CNC2, a 10Mb region in the 2p16 locus, in CNC1-negative patients. Nevertheless, the genes residing in that region remain unknown.6
Molecular pathways
The PKA heterotetramer consists of two regulatory (R) and two catalytic (C) subunits. When cAMP binds to the R subunits, the C subunits dissociate from the R subunits and thus phosphorylate many downstream factors. PRKAR1A defects lead to the loss of the regulatory subunit's function, leading to unrestrained catalytic activity and subsequent unrestrained cell proliferation and tumour formation.6 ,7
Most recently, components of the complex have been associated with defects of other PKA subunits, such as the catalytic subunits PRKACA (adrenal hyperplasia) and PRKACB (pigmented spots, myxomas, pituitary adenomas).6
Costello syndrome (CS) and cardiofaciocutaneous syndrome (CFCS)
Genetics
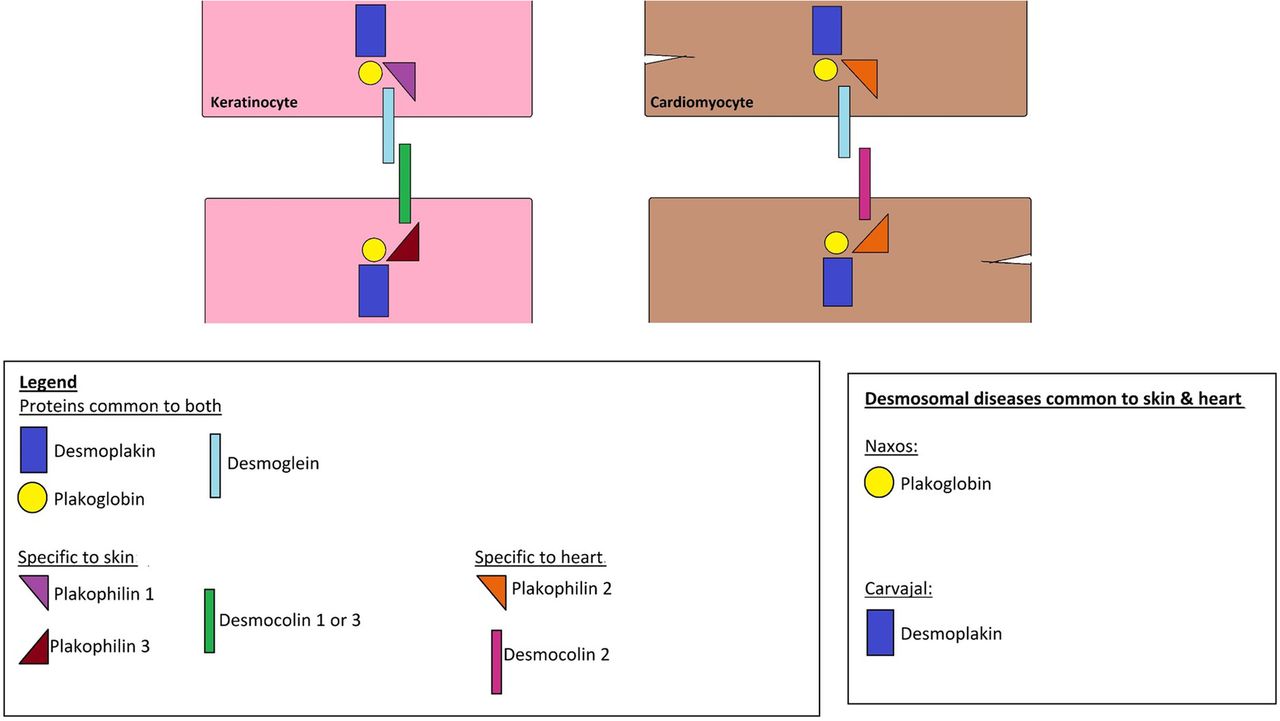
CFCS and CS are clinically related developmental disorders that belong to the family of RASopathies, a group of clinically and genetically related syndromes that are caused by mutations in the RAS/MEK/ERK signalling pathway (figure 2). CS is caused by de novo germline missense mutations in the HRAS proto-oncogene on chromosome 11p13.3. On the other hand, CFCS is a heterogeneous disorder that encompasses mutations in BRAF, MEK 1, MEK 2 and KRAS encoding proteins downstream of RAS.8

The RAS-MEK-ERK pathway represents a chain of protein signalling pathway that responds to a mitogen binding to a cell surface receptor. This chain of protein communications occurs through sequential protein phosphorylation and ends up by affecting the nuclear DNA transcription leading to different cellular changes. Abnormalities of certain factors along the pathway can lead to cardiocutaneous syndromes.
Costello syndrome
Cutaneous manifestations
The skin of patients with CS is usually velvety soft, with excessive wrinkling and deep palmoplantar creases. Hyperpigmented lesions such as diffuse or localised hyperpigmentation, nevi and acanthosis nigricans are often found. Whereas papillomas of the nasal areas or other sites are highly suggestive of CS, more typical features observed in this syndrome are sparse and curly hair as well as brittle and thin nails. Follicular hyperkeratosis (stacking of keratin in the openings of the hair follicles) of the arms, legs and face are commonly found, usually associated with sparse, slow-growing, curly hair and sparse eyebrows.9
Cardiac manifestations
Cardiac manifestations of CS include arrhythmias (usually atrial tachycardia), hypertrophic cardiomyopathy, pulmonary valvular stenosis and aortic dilation. These findings greatly contribute to a negative prognosis and are found in about 75% of patients.8
Other manifestations
Other manifestations have been reported in CS such as mental retardation, growth retardation, coarse facies, fetal overgrowth, friendly behaviour, rhabdomyosarcomas and other childhood malignancies and joint laxity.8
Molecular pathways
CS was first hypothesised to be due to an elastin disorder, as cultured skin fibroblasts were found to have dysfunctional elastin binding protein, which leads to a defect in the assembly of elastin fibres. The RAS/MAPK pathway is essentially involved in cell proliferation, differentiation, motility, apoptosis and senescence. The activation of this pathway enhances the proliferation of various types of cells, including melanocytes and keratinocytes.8 ,9 Furthermore, the c-KIT pathway, an activator of MITF (a regulator of melanin synthesis), may also activate the RAS/MAPK pathway. Nevertheless, the link between hyperactivation of the RAS/MAPK pathway and elastin fibre disruption is not well established until now.9
Cardiofaciocutaneous syndrome
Cutaneous manifestations
CFC is the RASopathy most frequently associated with sparse, curly and friable hair, with sparse or absent eyebrows and ulerythema ophryogenes (inflammatory, erythematous keratotic facial papules that may lead to scars, atrophy and alopecia). Patients with CFC typically have wavy or curly scalp hair, with sparse hair at the temples, poor hair growth, and sparse arm and leg hair (figure 3). About 90% of individuals with CFC have sparse or absent eyebrows due to ulerythema ophryogenes (erythematous with loss of follicles). Numerous acquired melanocytic nevi, not restricted to sun-exposed areas, are another striking feature of CFC. Other cutaneous manifestations observed in CFC include keratosis pilaris (follicular hyperkeratosis of the extremities and/or face), heat intolerance, hyperhidrosis and axillary body odour, dry skin, eczema, dystrophic fast-growing nails, generalised hyperpigmentation, acanthosis nigricans, hyperplastic nipples and creases on the fingertips.8–10

Cardiofaciocutaneous syndrome. (A) Keratosis pilaris affecting the face, with facial changes including saddle nose and low set ears. (B) Non-scarring hypotrichosis (decreased hair) over the scalp of an individual with cardiofaciocutaneous syndrome.
Cardiac manifestations
Approximately 75% of individuals with CFC have cardiovascular findings, the most common of which is pulmonary valvular stenosis, which may be accompanied by a dysplastic pulmonary valve. Pulmonary stenosis may occur in isolation, or concomitantly with other cardiac defects such as atrial septal defect or hypertrophic cardiomyopathy. Other cardiac defects associated with CFC include ventricular septal defect, mitral valve dysplasia, aortic coarctation and subaortic stenosis. These cardiac defects may appear early on in life, but a normal echocardiography does not preclude the possibility of developing cardiac abnormalities later on in life.9 ,10
Studies have found that MEK mutations may be associated with a higher likelihood of ventricular septal defects and dermatological manifestations, as opposed to BRAF mutations which may be more commonly associated with hypertrophic cardiomyopathy and atrial septal defects.11 ,12
Other manifestations
These include dysmorphic craniofacial features, namely macrocephaly, a tall forehead with bitemporal narrowing, increased facial width and depth, coarse facial features, ptosis, hypertelorism, downslanting palpebral fissures, epicanthal folds and a small chin.8 ,10
Neurocognitive abnormalities are universally present in CFC and range from mild to severe hypotonia, motor and speech delay, learning disability and behavioural dysfunction. Seizures are most frequently found in CFC as compared to the other RASopathies.8 ,10
Gastrointestinal and growth dysfunctions almost always include failure to thrive and poor growth in infancy, usually accompanied by severe feeding problems such as gastro-oesophageal reflux, suck/swallow dysfunction and oral aversion. These defects may first manifest as polyhydramnios in utero, which progresses to difficulty feeding after birth, as well as respiratory complications from oropharyngeal difficulties, namely choking and aspiration pneumonia.
Endocrine and musculoskeletal findings in CFC include short stature, global hypotonia, scoliosis and/or kyphosis, pectus deformities, joint hyperextensibility or contractures, and dysfunctional gait.8 ,10
Ophthalmological features of CFC occur in the majority of patients, namely strabismus, nystagmus, ptosis, refractive errors and optic nerve hypoplasia.10
External ear abnormalities such as low set, posteriorly rotated ears with upturned lobes, ear lobe creases, small postero-auricular indentations, ear pits and excess cerumen are also seen. Individuals with CFC have a peculiar dental phenotype characterised by malocclusion with bite, a high arched palate and high labial frenal attachment, as well as oral automatisms such as tongue thrusting, bruxism and open mouth posture.10
In summary, both CFCS and CS encompass numerous nevi, skin and joint laxity, pruritus and hyperhidrosis. Acanthosis nigricans, papillomas and loose, thick skin of the dorsum of the hands are characteristic of CS, whereas sparse eyebrows and dry hyperkeratotic skin are suggestive of CFCS.9
Ellis van Creveld syndrome (EVC)
EVC, also known as chondroectodermal dysplasia, or mesoectodermal dysplasia, is a rare autosomal recessive skeletal dysplasia. It is associated with mutations in the closely related EVC1 and EVC2 genes on chromosome 4p16, which are expressed in the heart, placenta, lung, liver and skeletal muscle.5 ,13
Cutaneous manifestations
Cutaneous manifestations of EVC are typically characterised by hidrotic ectodermal dysplasia of the nails, hair and teeth. Individuals have fine, sparse hair, hypoplastic fingernails, sausage-shaped digits and polydactyly.5 ,13
Cardiac manifestations
Congenital heart defects occur in 60% of individuals with EVC syndrome, and include single atrium, mitral and tricuspid valve defects, patent ductus arteriosus, ventricular and atrial septal defects, and a hypoplastic left heart. Cardiac defects seem to be the main determinant of longevity in this disease.5 ,13
Other manifestations
Short ribs, short limbs, disproportionate small stature with increasing severity from the proximal to the distal portions of the limbs, shortening of the middle and distal phalanges, polydactyly, dysplastic teeth and nails.5 ,13 ,14
Oral manifestations include malocclusion, labiogingival adherence and gingival hypertrophy, labiogingival frenulum hypertrophy, conical teeth, enamel hypoplasia and hypodontia.5 ,13
Genetics and molecular pathways
The EVC gene encodes a protein whose function is unclear; however, it seems to be important for normal growth and development, especially of the bones and teeth. It helps in the regulation of a Sonic Hedgehog signalling pathway that plays important roles in cell growth, specialisation and patterning of body parts.15 ,16
The H syndrome
Cardiac, cutaneous and other manifestations
The H syndrome is a recently described autosomal recessive systemic genodermatosis, characterised by multiple H's (figure 4):
Hyperpigmentation and hypertrichosis: indurated patches on different parts of the body—heart anomalies
Hearing loss: sensorineural
Hypogonadism: hypergonadotropic hypogonadism, gynaecomastia
Hepatosplenomegaly
Low height
Hyperglycaemia: occasionally; could lead to insulin-dependent diabetes mellitus
Hallux valgus; and camptodactyly: fixed flexion contractures of the toes and proximal interphalangeal joints.17

H syndrome. (A) Hyperpigmentation and hypertrichosis: indurated (hard) patches on different parts of the body. (B, C) Hallux valgus and camptodactyly: fixed flexion contractures of the toes and proximal interphalangeal joints.
Genetics and molecular pathways
H syndrome is caused by mutations in the SLC29A3 (solute carrier family 29 (nucleoside transporters), member 3) gene, which encodes hENT3, a member of the ENT family. hENT3 acts as an essential nucleoside transporter in the salvage pathway of nucleotide synthesis in cells that lack de novo synthesis.18
Leopard syndrome (LS)
LS refers to an acronym based on the major features of the disorder:
MultipleLentigines
ECG conduction abnormalities
Ocular hypertelorism
Pulmonary stenosis
Abnormal genitalia
Retardation of growth
SensorineuralDeafness
It is also known as multiple lentigines syndrome, cardiocutaneous syndrome, lentiginosis profusa, Moynahan syndrome and progressive cardiomyopathic lentiginosis.19
Cutaneous manifestations
LS is characterised by multiple lentigines, which are flat, black-brown macules dispersed mainly on the face, neck and upper trunk, but sparing the mucosae. They generally appear at the age of four to five and increase into the thousands until puberty, independently of sun exposure. Patients with LS also exhibit premature skin wrinkling as well as café au lait spots.19 ,20
Cardiac manifestations
The most common heart defects associated with LS are ECG anomalies (75% of patients) and progressive conduction anomalies (23% of patients). ECG abnormalities include left or biventricular hypertrophy (often with Q waves), prolonged QTc and repolarisation abnormalities. Conduction defects include a superiorly oriented mean QRS axis in the frontal plane, even with a normal cardiac structure.19
While pulmonary valve stenosis (with or without dysplasia) and mitral valve prolapse may be observed, the most frequent cardiac anomaly reported is hypertrophic cardiomyopathy. The latter may be life-threatening, and its onset usually precedes the lentigines, but may worsen in parallel with their appearance.19 ,20
Other manifestations
Typical findings in LS include facial dysmorphisms that become more striking with age, such as hypertelorism, flat nasal bridge, ptosis, deep nasolabial folds and dysmorphic ears.
Common findings include growth retardation, a broad chest, pectus deformities, bilateral cryptorchidism, hypospadias, genital hypoplasia, hypotonia and delayed psychomotor development. Less frequent are reports of prognathism, winging of the scapulae, scoliosis and joint hyperextensibility.19
Genetics and molecular pathways
LS may be sporadic or inherited in an autosomal dominant fashion with full penetrance. It is mainly caused by a missense mutation in the PTPN11 gene on chromosome 12q24.1. This gene encodes SHP-2, a cytoplasmic signalling transducer downstream of multiple receptors for growth factors, cytokines and hormones, with a particular role through the MAPK pathway. Recently, mutations in RAF121 and BRAF19 ,22 genes have also been described in association with an LS phenotype.
Noonan syndrome (NS)
NS is another developmental disorder belonging to the RASopathies. Eleven of the 15 different genes affecting the RAS-MAPK pathway have been found to be involved in NS, half of which are mutations in PTPN11.
Cutaneous manifestations
These include keratosis pilaris, hyperkeratosis, hair abnormalities, pigmented nevi, lentigines and café au lait spots.23 ,24
Cardiac manifestations
The most common congenital heart defect is pulmonary valve stenosis with dysplastic leaflets (50–62%). Twenty per cent of patients are found to have hypertrophic obstructive cardiomyopathy with asymmetrical septum hypertrophy. Other less common cardiac findings include atrial and ventricular septal defects, persistent ductus arteriosus, mitral valve anomalies and atrioventricular canal defect associated with subaortic obstruction. Electrocardiographic abnormalities include wide QRS complexes with a predominantly negative pattern in left precordial leads. Some patients may be found to have left axis deviation and giant Q waves.23 ,25
Other manifestations
These include short stature, unusual pectus deformity, failure to thrive, mild mental retardation, bleeding abnormalities and skeletal defects. Typical facial features commonly reported encompass hypertelorism, ptosis, downslanting palpebral fissures, a triangular face, low-set, posteriorly rotated ears and a broad forehead. Patients may also be found to have macrocephaly, a webbed neck and cryptorchidism.23 ,24
Genetics and molecular pathways
NS is caused by autosomal dominant mutations in PTPN11, SOS1, RAF1, KRAS, NRAS and BRAF genes. Most cases of NS result from mutations in one of three genes: PTPN11 (50%), SOS1 (10–15%) or RAF1 (5–10%). All these genes play important roles in signalling pathways involved in cell division, movement, differentiation and thus proper tissue development.23 ,24
Pseudoxanthoma elasticum (PXE)
PXE, also known as Gröenblad-Strandberg syndrome, is characterised by fragmentation of elastic fibres due to aberrant mineralisation of soft connective tissue, primarily in the skin, cardiovascular system and eyes.26 It affects women more than men, and rarely presents at birth; its manifestations become most prominent in the second and third decades of life.27–29
Cutaneous manifestations
The first signs of PXE consist of small (1–5 mm), asymptomatic, yellowish or skin-coloured papules, distributed in a reticular pattern initially on the lateral and posterior neck. Later, these lesions coalesce into larger plaques and involve flexural areas such as the axillae, inguinal region, antecubital and popliteal fossae, and periumbilical area (figure 5).30 These lesions may even involve the mucosa of the oral cavity and inner lower lip, as well as the genital area. The skin thus becomes lax, redundant and wrinkled.27 ,31

Pseudoxanthoma elasticum. Multiple skin coloured to yellowish papules coalescing into plaques over neck and forearm in a young individual admitted for cerebral haemorrhage.
Cardiovascular manifestations
Cardiovascular involvement in PXE is mainly characterised by reduced peripheral pulse, angina pectoris, hypertension and intermittent claudication. Patients with PXE are at risk of developing premature atherosclerosis and dyslipidaemia, with early myocardial infarcts and cerebrovascular accidents. Patients may also report haematemesis and melaena, which are signs of gastrointestinal haemorrhage. The faulty mineralisation of PXE mainly affects the internal elastic lamina, media and adventitia of medium-sized arteries and the aorta, as well as the endocardium, pericardium, connective tissue of the myocardium and coronary arteries.27 ,32 ,33
Other manifestations
Ophthalmological involvement constitutes a significant portion of the manifestations seen in patients with PXE. Individuals most commonly present with characteristic angioid streaks of the retina, which may either be discovered incidentally on routine ophthalmological examination, or in association with retinal haemorrhage. Angioid streaks result from fractures in Bruch's membrane, an elastin-rich sheath within the retina. Owing to fragmentation of this membrane, the retinal blood vessels break, consequently leading to neovascularisation. The most frequent cause of morbidity and disability in PXE is reduced visual acuity from macular haemorrhage and disciform scarring. These findings typically manifest in the third decade of life. In contrast, a peau d'orange appearance of the fundus typically appears between adolescence and the second decade of life.27 ,34 ,35
Genetics and molecular pathways
PXE is an autosomal recessive disease caused by mutations in the ABCC6 (ATP-binding cassette subfamily C member 6) gene, on chromosome 16p, which encodes a transmembrane ATP driven anion transporter. Studies have shown that a high intake of dairy products rich in calcium and phosphate during childhood and adolescence, or intake of aluminum hydroxide (a phosphate binder), may play a role in the pathological mineralisation underlying PXE.36 ,37
Currently, two hypotheses exist to explain the pathomechanism behind PXE: the metabolic and the cellular hypotheses. The metabolic hypothesis states that the lack of ABCC6 activity in the liver leads to a deficiency of antimineralisation factors which normally prevent precipitation of calcium phosphate complexes and thus aberrant mineralisation.38 The cellular hypothesis postulates that the absence of ABCC6 expression leads to the accumulation of minerals in soft connective tissues, mainly fibroblasts, thus disturbing the normal cell homeostasis, with subsequent local mineralisation.39 ,40
Naxos–Carvajal syndromes
Naxos syndrome belongs to a spectrum of related disorders, which is characterised by arrhythmogenic cardiomyopathy, peculiar woolly hair and palmoplantar keratoderma (figure 6). The WHO has considered it to be the recessive form of arrhythmogenic right ventricular dysplasia/cardiomyopathy (ARVD/C).41

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
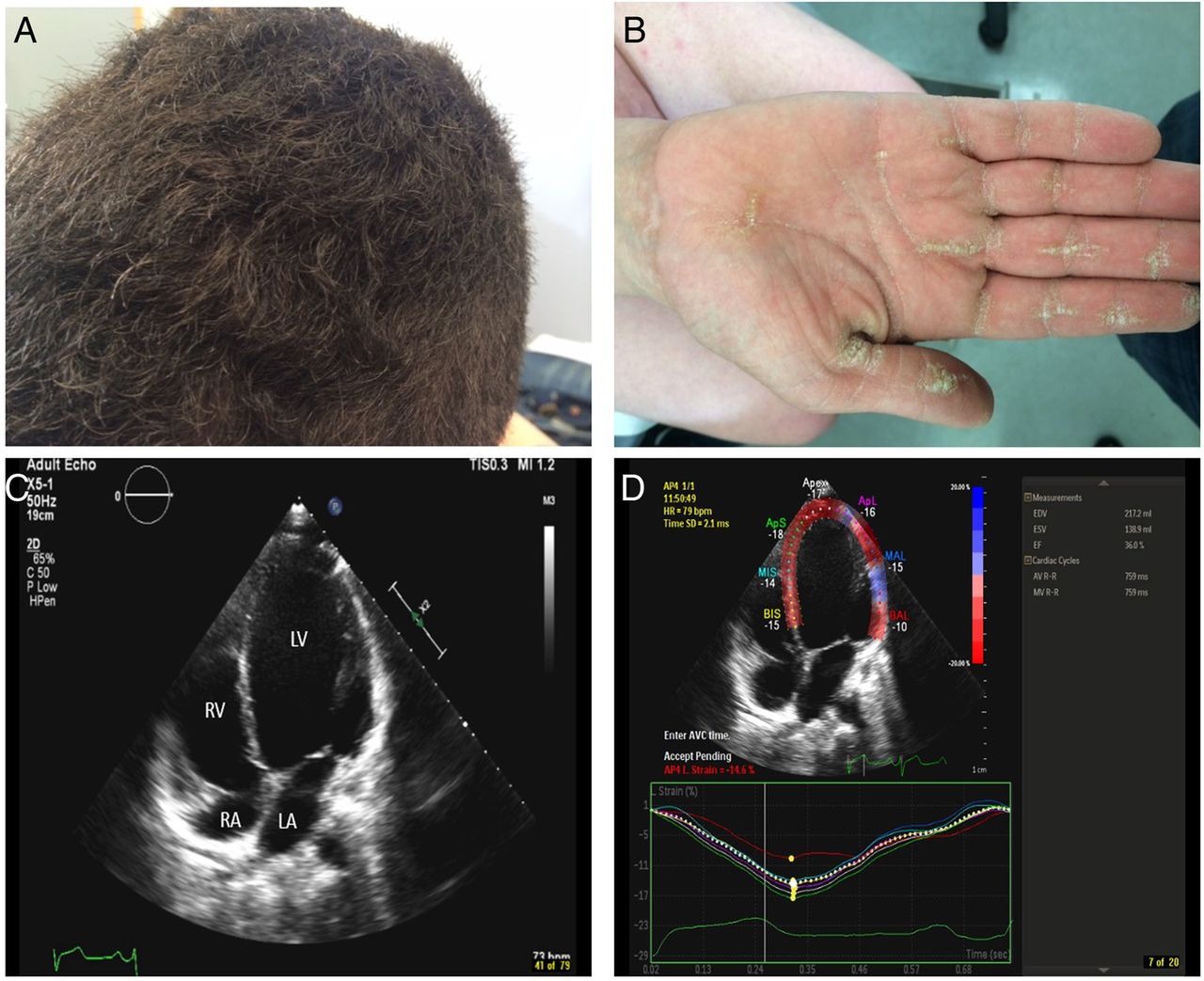
Carvajal syndrome. (A) Woolly hair (curly hair which at times could be subtle but could be detected when comparing the hairs to members of the same family, such as the case here. (B) Palmar keratoderma. Striate type of keratoderma (longitudinal thickening of the palmar skin) which in a setting of woolly hair should raise the suspicion for Naxos/Carvajal syndrome. (C) Apical four-chamber view demonstrates a dilated LV cavity with a spherical appearance with an ejection fraction of around 28%. LA, left atrium; LV, left ventricle; RA, right atrium and RV, right ventricle. (D) Peak systolic longitudinal strain measurement by speckle tracking with strain curves reveals a decrease in the global longitudinal strain of the left ventricle (average strain of—14.6% of all segments; normal for age is –19%).
Carvajal syndrome is a variant of Naxos syndrome that is characterised by younger age at presentation and more prominent left ventricular involvement, as well as clinical overlap with dilated cardiomyopathy.42
Cutaneous manifestations
The cutaneous findings of Naxos–Carvajal syndrome vary with age; woolly hair appears since birth, whereas palmoplantar keratoderma develops during the first year of life as infants begin using their hands and feet (figure 6A, B). There have been several case reports of leukonychia, as well as hypo/oligodontia in patients with this syndrome.41–43
Cardiac manifestations
Symptomatic cardiomyopathy, on the other hand, manifests by adolescence with 100% penetrance. Patients typically present with syncope and/or sustained ventricular tachycardia with left bundle branch block. Conversely, sudden death may be the first presenting symptom of the disease. Abnormalities in repolarisation and/or depolarisation on a restingECG, and structural/functional right ventricular abnormalities on echocardiography have been reported in all patients. The right ventricular myocardium, mainly the subepicardial and mediomural layers, is typically replaced with fibro-fatty tissue. Half of the patients develop progressive heart disease that may involve both ventricles. These cardiac findings are a significant cause of mortality in Naxos disease with left-sided dilated cardiomyopathy characteristic of Carvajal syndrome with mutations in desmoplakin (figure 4C, D).41 ,44 ,45
Genetics and molecular pathways
Naxos and Carvajal syndromes are autosomal recessive disorders caused by mutations in the plakoglobin and desmoplakin genes, respectively, on chromosome 17q21. Cardiac myocytes are connected by complex intercellular contact sites called intercalated discs, which contain three different types of intercellular junctions. Adherens junctions and desmosomes enable synergistic contraction via mechanical coupling, whereas gap junctions allow a rapid spread of action potentials via electrical coupling. Plakoglobin is a component of both adherens junctions and desmosomes. Plakoglobin and desmoplakin are intracellular proteins that anchor desmosomes to desmin intermediate filaments. Plakoglobin also contributes to interlinking adherens junctions with the actin cytoskeleton, and is involved in apoptosis. Mutated plakoglobin thus leads to defective myocardial mechanical coupling, which interrupts the contiguous chain of cell adhesion, especially under mechanical stress or stretch. This leads to cell death and progressive fibro-fatty replacement, which furthermore provides a nidus for re-entry ventricular arrhythmias. Desmosomes are also abundant in the epidermis, providing an explanation for the cutaneous manifestations seen in these diseases.41 ,43 ,46
Ehlers-Danlos syndromes (EDS)
EDS are a group of heterogeneous disorders of connective tissue, characterised by varying degrees of fragility of the skin, blood vessels, ligaments and internal organs. The Villefranche classification categorises EDS into six subtypes, the most common of which are the classic, hypermobility and vascular types. In contrast, the kyphoscoliosis, arthrochalasis and dermatosparaxis types are rare. These categories generally differ in clinical findings, mode of inheritance and biochemical/molecular findings; nevertheless, a significant overlap does exist.47 ,48
Cutaneous, cardiac and other manifestations
EDS are characterised by varying degrees of skin hyperextensibility, joint hypermobility, delayed wound healing with atrophic cigarette paper scarring, easy bruisability and generalised fragility of the soft connective tissues. The skin of patients with EDS is smooth, velvety and hyperelastic (extends easily and recoils back after release), as opposed to the skin in cutis laxa syndromes. Skin hyperextensibility is a characteristic of all EDS subtypes except the vascular type, in which the skin is thin and transparent, thus revealing the venous pattern over the chest, extremities and abdomen. Furthermore, this subtype of EDS warrants special attention due to its association with life-threatening complications, such as arterial rupture (with or without a preceding aneurysm), arteriovenous fistulae and dissections. In those with the ‘acrogeric’ form, the skin over the distal extremities has an aged, thin and atrophic appearance.48
Dermatosparaxis, EDS type VIIC, is a peculiar subtype characterised by striking joint laxity, extremely soft, fragile and extensible skin, easy bruisability and mild hirsutism. Other associated features include short fingers, umbilical hernia, small chin, eyelid oedema, blue sclerae and delayed closure of the fontanelles.48
Genetics and molecular pathways
Most forms of EDS have been linked to mutations in genes encoding collagens or collagen-modifying enzymes involved in the post-translational processing of collagen. Collagen type V has been found to be mutated in the classic subtype (COL5A1 more often than COL5A2), as opposed to the mutated collagen type III in the vascular subtype and the collagen type I in the arthrochalasis, kyphoscoliosis and dermatosparaxis variants.
In addition, mutations in tenascin-X, a non-collagenous protein, leading to a complete lack of serum tenascin-X, have been associated with an autosomal recessive phenotype that greatly resembles classic EDS but without atrophic scars. Tenascin-X is an extracellular matrix protein that regulates collagen deposition by fibroblasts.
The Dermatosparaxis subtype is a recessively inherited disorder that results from absence of the activity of procollagen N-proteinase (now known as ADAMTS2). ADAMTS2 encodes an enzyme that cleaves the N-terminal propeptide from type I procollagen.48 ,49
Marfan syndrome (MFS)
MFS is a systemic disorder of the connective tissue that affects the cardiovascular, ocular and skeletal systems. It affects both sexes equally and demonstrates an autosomal dominant inheritance.48
Cutaneous manifestations
Cutaneous involvement constitutes part of the minor criteria for MFS. Findings include striae distensae independent of marked weight gain or pregnancy.48
Cardiovascular manifestations
Cardiovascular pathology accounts for the leading causes of mortality and morbidity in patients with MFS. Such findings include aortic root dilation, dissection and rupture. The majority of fatal events occur in early adult life; as a result, timely recognition and appropriate intervention can greatly increase mean survival age to 72 years. Other cardiovascular findings comprise mitral valve dysfunction; myxomatous valve thickening may lead to prolapse, insufficiency and calcification. In contrast, aortic valve insufficiency is the result of aortic root dilation.50
Other manifestations
Patients with MFS have striking long bone overgrowth that leads to disproportionately long limbs and anterior chest deformities due to rib overgrowth. Other major features of this syndrome include arachnodactyly, elbow contractures, scoliosis, spondylolisthesis, downslanting palpebral fissures, enophthalmia, retrognathia and a high arched palate with tooth crowding. Patients typically present with joint hypermobility and its consequent ligamentous injury, dislocations, chronic joint pain and premature arthrosis. Ocular lens dislocation (ectopia lentis), high myopia, retinal detachment, cataract and glaucoma are common ophthalmological manifestations of the disease. Recurrent inguinal or surgical hernias, pneumothorax, pulmonary emphysema and restrictive lung disease have also been reported.48
Genetics and molecular pathways
FBN1 has been identified as the causal gene, but recent investigations have implicated perturbed cytokine signalling as a culprit. FBN1 gene encodes fibrillin-1, an important extracellular matrix glycoprotein whose monomers aggregate to form microfibrils, which in turn associate with elastin to form elastic fibres. However, recent studies on mouse models have shown that deficiency in fibrillin-1 leads to increased activation of TGF-β and subsequent perturbation of downstream cascades.51 ,52
Cutis laxa
Cutaneous manifestations
Cutis laxa is clinically defined by loose and saggy skin that demonstrates reduced elasticity and resilience. The skin appears furrowed, especially in the neck, axillae and groin. The face has a ‘droopy’ appearance with ptosis and drooping cheeks. In contrast to Marfan and Ehlers-Danlos, the skin in cutis laxa does not display hyperelasticity. In newborns with severe cutis laxa, the elastic fibres can be essentially absent. In many cases with late-onset cutis laxa, although present, the elastic fibres are fragmented and lack contiguous functionality.53
Cardiac manifestations
These include peripheral pulmonary artery stenosis, vascular malformations and supravalvular aortic stenosis (rarely). The most common cause of death in patients with cutis laxa is cardiorespiratory failure from complications of pulmonary emphysema.53
Other manifestations
These mainly include early childhood-onset pulmonary emphysema, umbilical and inguinal hernias, gastrointestinal and vesicourinary tract diverticuli.53
Genetics and molecular pathways
Cutis laxa is caused by mutations in the elastin gene. A limited number of mutations have been identified in the FBLN5 gene encoding fibulin-5, a microfibrillar component of the elastic fibres. Of note, mutations in the LTBP4 gene have been found to be associated with an early lethal phenotype and severe gastrointestinal and genitourinary malformations. The LTBP4 gene encodes latent TGF-β binding protein 4, whose absence in the extracellular matrix causes increased TGF-β activity, leading to defective elastic fibre assembly in affected tissues.54
The list of genetic cardiocutaneous syndromes goes beyond what we have already mentioned and additional less common syndromes are listed in table 1.
Other cardiocutaneous syndromes: genetics, cutaneous and cardiac manifestations, and other key findings
To conclude, in this review, we aim to highlight clinical clues that may alert dermatologists, cardiologists, internists, as well as general practitioners to scrutinise certain cutaneous lesions that may sometimes seem to be superficial and trivial. Further investigations may uncover more serious cardiac diagnoses associated with these cutaneous lesions that would have otherwise been hidden, undetected diseases with deleterious and fatal consequences. These are an example of how internal processes can have cutaneous manifestations and present with dermatological clues that may be missed even by skilled physicians, thus highlighting the importance of intelligent recognition, early detection and adequate treatment.
References
Footnotes
GN and MK These authors contributed equally to the work.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement No additional data are available.