Article Text

Abstract
Objective Functional (or secondary) mitral regurgitation (FMR) is associated with greater morbidity and worse outcomes in patients with congestive heart failure (CHF) and cardiomyopathy. The Carillon® Mitral Contour System® is a coronary sinus-based percutaneous therapy to reduce FMR. We evaluated the safety and efficacy of a modified version of the Carillon device in the treatment of patients with cardiomyopathy and FMR.
Methods 36 patients with CHF, depressed left ventricular function (ejection fraction <40%) and at least moderate FMR underwent the Carillon device implant.
Results There was 1 major adverse event within 30 days—a death (not device related)—occurring 17 days after the implant. Reductions in FMR and improvements in functional class and 6 min walk tests were seen, similar to prior studies. Device fractures in the high strain region of the proximal anchor (seen in prior studies) were not seen in this study.
Conclusions The modified Carillon device was associated with improvements in clinical and echocardiographic parameters in treating patients with FMR, while successfully addressing the issue of anchor fracture. This version of the Carillon device will be used in a blinded randomised trial of symptomatic patients with FMR.
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Key questions
What is already known about this subject?
It is unclear what treatments improve outcomes with functional mitral regurgitation. Two prior studies using the Carillon device provided evidence for safety and efficacy, but asymptomatic fractures of the previous iterations of the device were seen.
What does this study add?
This study confirms the safety profile of the Carillon device, without having the device fractures seen in prior studies. This study further supports the efficacy of the device, with improvements similar to those seen in prior studies.
How might this impact on clinical practice?
The Carillon device is one of the first devices with the Conformité Européene mark of approval to percutaneously treat mitral regurgitation that is commercially available (in certain European countries). The device iteration used in TITAN II is being studied in a blinded randomised trial being carried out in several European countries and in Australia.
Secondary, or functional, mitral regurgitation (FMR) is a common condition in patients with dilated cardiomyopathy in the setting of congestive heart failure.1–7 Patients with heart failure and FMR have worse haemodynamic characteristics and clinical outcomes than patients without FMR.1 ,2 ,5–14 Two previous studies of the Carillon Mitral Contour System, AMADEUS and TITAN trials, have shown improved clinical and echocardiographic parameters when used to treat symptomatic patients with FMR, with an acceptable safety profile, given the high risk of patients being treated.15–17 However, clinically benign wire-form fractures were seen next to the proximal anchor locking mechanism, at high strain locations on the Carillon device. The TITAN II study was performed to evaluate the safety and integrity of a modified Carillon Mitral Contour System with design changes to improve the fatigue resistance of the device (figure 1).

The top device is the XE2, used in TITAN. The bottom device is the modified XE2 (mXE2), used in TITAN II. Among other changes, the XE2 has a ‘ski-slope’ in the wireforms next to the end, not seen in the mXE2 (thin arrows). There is a little distance between the crimping tube of the proximal anchor and the locking loop of the mXE2 (thick arrow), not seen in the XE2. These helped to reduce the strain in the wireforms of the proximal anchor next to the end of the device, where the majority of the fractures occurred in TITAN.
Methods
The study was a prospective, single-arm, multicentre safety study undertaken at five centres in Germany, Poland and France.
The primary end point was the rate of major adverse events at 30 days, defined as: death, myocardial infarction, cardiac perforation necessitating intervention, device embolisation or the occurrence of surgery or percutaneous intervention related to the device. Secondary safety end points included mortality and major adverse event rates up to 12 months. Secondary efficacy end points include evaluation of mitral valve competence based on quantitative echocardiographic parameter and mitral regurgitation (MR) grade changes over 12 months. Secondary functional improvements include evaluation of the New York Heart Association (NYHA) classification 6 min walk improvement over 12 months follow-up.
Study design
The study design, including inclusion and exclusion criteria, was similar to the AMADEUS and TITAN trials,15 ,16 with minor modifications to the device in each study. There were only minor differences between the studies. For example, patients with atrial fibrillation were excluded from AMADEUS but included in TITAN and TITAN II. Patients were included if they had symptomatic congestive heart failure at least NYHA class 2 who were stabilised on heart failure medications for at least 1 month. In order to qualify, patients had to have 2+ or greater (at least moderate) MR, a 6 min walk test (6MWT) of 150–450 m, as well as high-risk features, including a dilated left ventricle (left ventricular end-diastolic diameter (LVEDD)>55 mm), and depressed left ventricular function with a left ventricular ejection fraction <40%. Patients were excluded if the aetiology of heart failure was not due to ischaemic or idiopathic dilated cardiomyopathy, or if the MR was due to a degenerative aetiology. Additional exclusions included the presence of a cardiac resynchronisation therapy (CRT) lead in the coronary sinus, severe renal insufficiency (creatinine>2.2 mg/dL), need for revascularisation or percutaneous coronary intervention within the prior 30 days, a myocardial infarction or coronary artery bypass graft surgery within the prior 3 months, or use of mechanical or inotropic support within the prior 30 days.
Patients underwent baseline screening, including echocardiography, a 6MWT and assessment of their baseline clinical status by NYHA classification, as well as evaluation for medication use, and baseline chemistries. Major adverse events were reviewed by an independent Data Safety and Monitoring Board.
The Carillon Mitral Contour System procedure has been previously described.15 ,16 The procedure can be performed under general anaesthesia or conscious sedation, depending on the preference of the operator, and on whether transoesophageal or transthoracic echo was used during the procedure. A 9 French sheath is placed in the right internal jugular vein and standard arterial access for coronary arteriography is obtained. From the internal jugular vein, the coronary sinus is accessed with a wire and a 7 French multipurpose or Amplatz diagnostic or guiding catheter inside of a proprietary 9 French curved delivery catheter. A marker catheter is placed inside the delivery catheter after it is placed in the great cardiac vein, to be used as a measuring device. Measurements are made of the venous dimensions, including vein diameters at the sites of anticipated anchor placements, as well as usable vein length.
The device is composed of two self-expanding nitinol anchors, with a curvilinear segment connecting them (figure 1), and is incorporated into a dedicated delivery system. The distal anchor is unsheathed in the great cardiac vein and then locked. Tension is applied to the system by pulling back on the delivery system at the level of the internal jugular vein. Coronary angiography is performed to evaluate for coronary artery compression. If the coronary artery is adequately patent, the proximal anchor is then unsheathed and locked. Echocardiography (either transoesophageal or transthoracic) is performed to evaluate improvement in MR. If everything appears acceptable, the device is released, the delivery system retrieved and the procedure concluded.
If there is coronary artery compromise, slipping of the distal anchor on applying tension or placement at the incorrect location, the tension can be released and the device recaptured and removed, as necessary, prior to releasing the device. An additional attempt with a new device may be tried, perhaps at a more proximal location if coronary compromise was an issue.
Patients receiving implants were followed at 1, 6 and 12 months intervals for clinical and echocardiographic parameters. Clinical parameters included assessment of NYHA classification, 6MWTs, changes in medications and hospitalisations. Echocardiographic parameters included quantitative assessments of MR by an independent core echo laboratory (Brigham and Women's Hospital, Boston, Massachusetts, USA), as well as measurements of left ventricular, mitral annular and left atrial dimensions, and ejection fraction. Cinefluoroscopy was performed of implanted devices at discharge, 1, 3, 6 and 12 months after implantation to evaluate device integrity.
Statistical analysis
Continuous variables are reported as mean and SDs. Categorical variables are presented as n/N (%).
Repeated measures analysis of variance (ANOVA) was used to estimate means of each measure at each visit to account for repeated measures per participant; a compound symmetry working correlation structure was used. p Values for the assessment of a significant impact of time (visit) are based on F tests from the ANOVA models. Similar analysis of covariance (ANCOVA) models were used to estimate the change from baseline at each visit. The dependent variable was change from baseline, while visit and baseline value of the measure were included as covariates. A compound symmetry working correlation structure was used to model the covariance between repeated measures per participant. p Values for the assessment of change from baseline are based on t-tests from the ANCOVA models. All statistical testing was carried out using a two-sided α=0.05 significance level.
Results
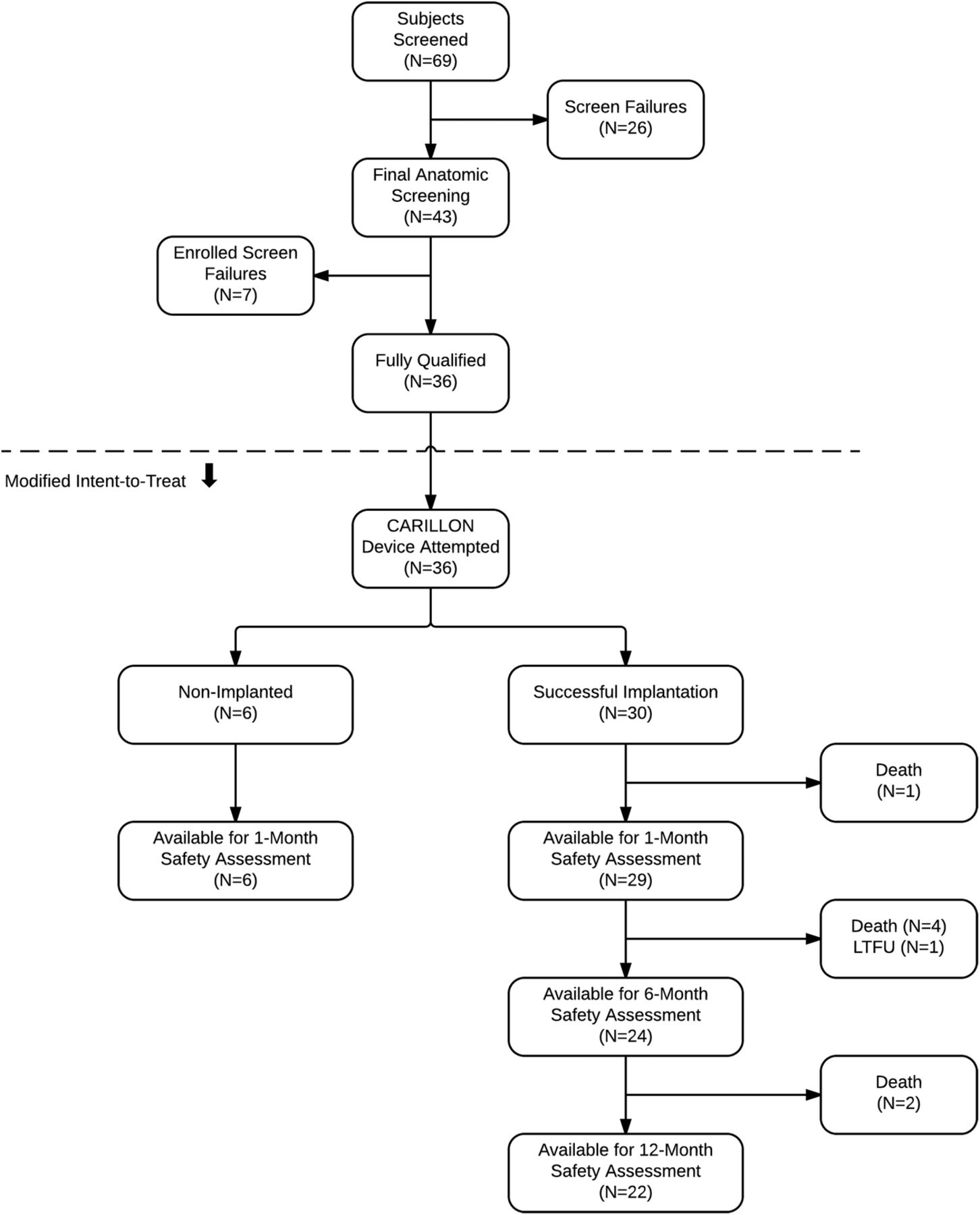
Sixty-nine patients were screened for inclusion in TITAN II. Of these, 26 were screen failures, as they did not meet entry inclusion/exclusion criteria (figure 2). Of the 43 consented patients, there were 7 enrolment failures, due to findings at the time of the procedure, including unanticipated coronary artery disease requiring revascularisation (n=4), presence of a left atrial clot (n=1) or venous dimensions not suitable for a Carillon implantation (n=2). Thus, there were 36 patients who underwent the Carillon procedure.

Patient disposition in TITAN II. LTFU, lost to follow up.
Baseline characteristics of the intent-to-treat cohort are shown in table 1. An ischaemic aetiology for the cardiomyopathy was seen in 58% of patients and 47% had chronic atrial fibrillation. The echo core laboratory assessed mean ejection fraction was 34.0±10.1%, with a mean LVEDD of 62.3±8.2 mm.
Baseline characteristics of enrolled patients
Six patients (17%) had uncomplicated, unsuccessful procedures with no Carillon implant deployed. Coronary artery compromise was the reason for device removal in all six cases. Three of these patients had single implant attempts, two had two device attempts and one had three attempts. Three of these patients had an ischaemic aetiology and the other three had non-ischaemic cardiomyopathy. These non-implanted patients were followed for 30 days, for safety events only.
Thus, a total of 30 patients successfully received permanent Carillon implants (83%). Twenty-two patients were successfully implanted with a single attempt (73%). Seven required two attempts and one required three attempts.
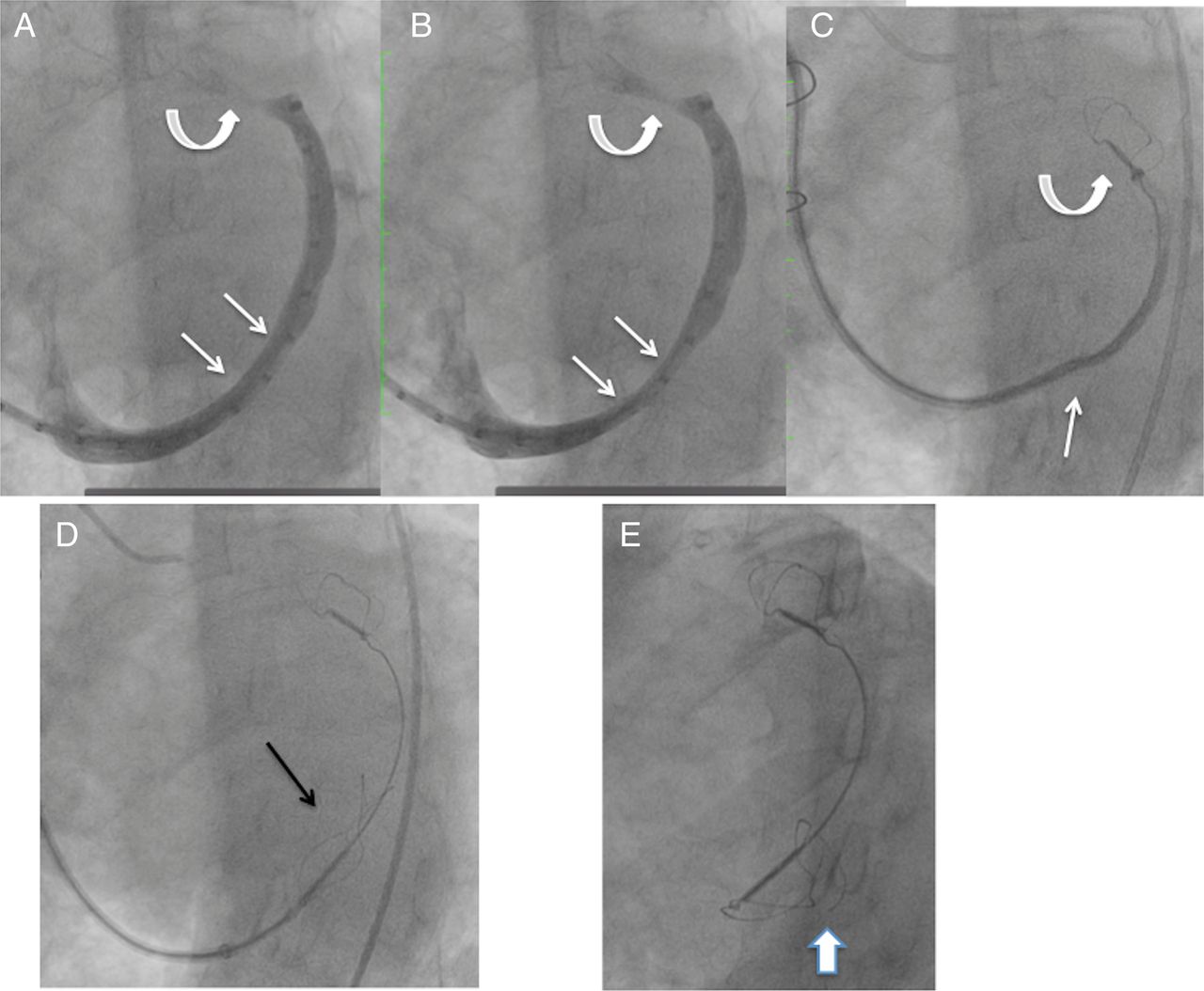
One patient had coronary angiography performed after placement of both anchors, finding a partial compromise of a small calibre, diseased atrioventricular branch of the circumflex coronary artery, which caused chest pain. The proximal anchor was recaptured, resolving the coronary compression and chest pain, but it was not possible to recapture the distal anchor. This was due to the distal anchor placement in a very small distal aspect of the great cardiac vein, beyond a sharp angle. Eventually, the recaptured proximal anchor, now distorted, was redeployed in the middle of the great cardiac vein, without tension in the system (figure 3). Thus, there were no complications from the procedure, but the device was not appropriately placed.
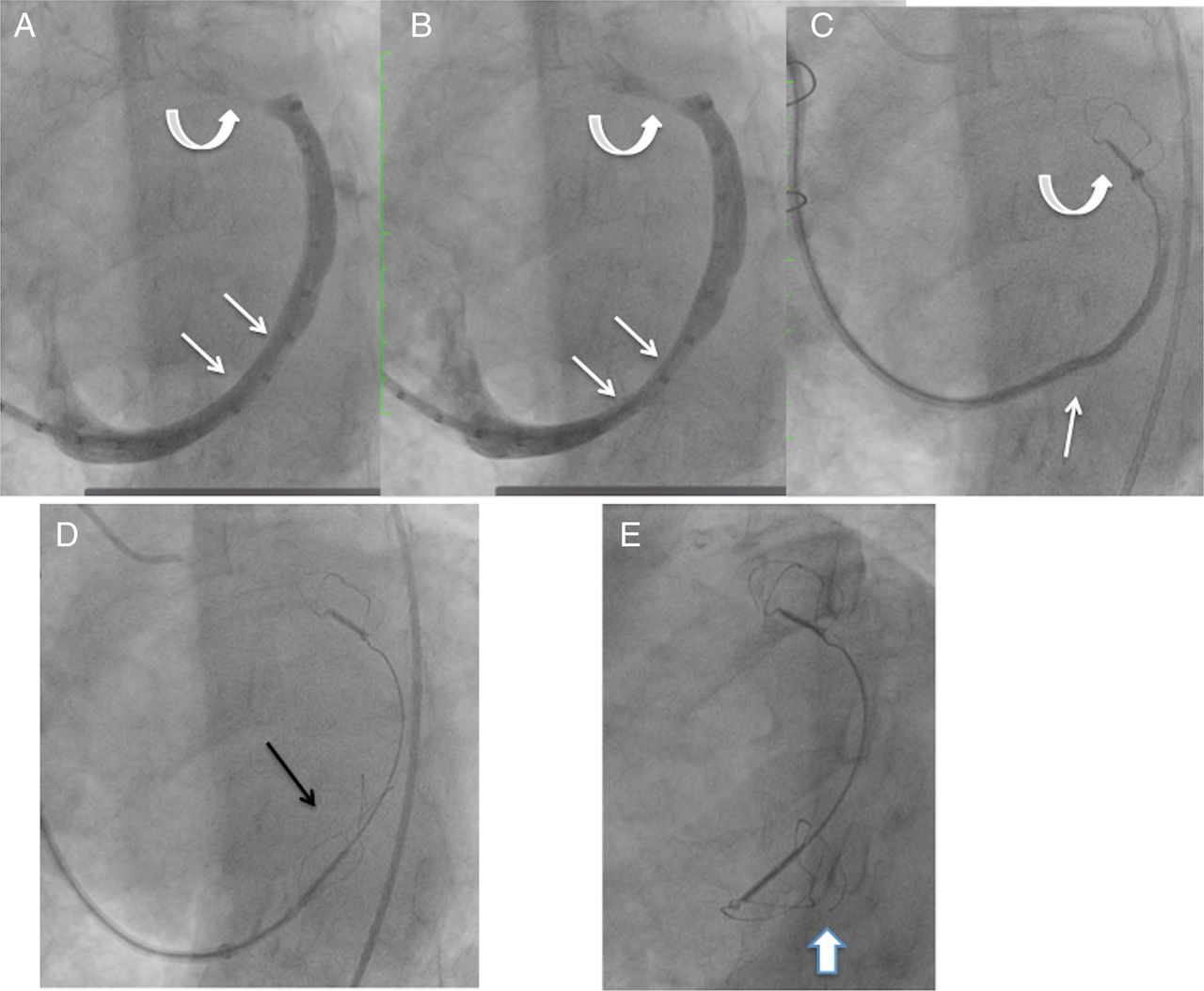
Venogram and image of the Carillon device during attempted recapture and as left in situ, prior to release, and at 3-month follow-up. (A and B) Coronary sinus/great cardiac vein venogram showing a dynamic component of the vein at site where the proximal anchor was eventually deployed (straight arrows). Curved arrows show the small calibre vein site where the distal anchor was placed. (C) Distal anchor in the small calibre portion of the vein (curved arrow), during attempted recapture. The straight arrow shows buckling of the catheter. The distal anchor appears lower than the small calibre portion of the vein as it is being pulled down due to the tension in the system. (D) The tethered Carillon device in position for release, after the proximal anchor had previously been placed, locked and recaptured, and now redeployed without tension in the system, with the location at the site of the dynamic compression seen in A and B. Arrow points to proximal anchor. (E) Follow-up cine 3 months later shows fracture of the proximal anchor (arrow), at a different location than where fractures had been noted with the prior version of the device.
Implant success was 83% (30 of 36). In 7 of 36 cases (19%), coronary compromise limited device deployment, including the patient described above in whom a device was implanted, but not at the desired location of the proximal anchor.
The mean total fluoroscopy time and total procedure time (time from first sheath insertion until the last catheter was removed from the body) were 36±28 min (median 29 min) and 116±48 min (median 102 min), respectively, for the intention-to-treat population in which an implant attempt was made. The mean (and median) total implant time (time from the Carillon delivery catheter being introduced into the coronary sinus until the device was decoupled) for patients with successful implants was 75±40 min (median 65 min), with a mean fluoroscopy time of 33±27 min (median 28 min). The mean contrast usage was 186±93 mL, with a range of 50–500 mL.
Primary end point
There were no device-related major adverse events. There was one procedure-related (categorised as possibly related) major adverse event—a sudden death occurring 17 days after successful device implantation in a patient with ischaemic cardiomyopathy and atrial fibrillation. The patient was admitted to the hospital 13 days after a successful device implantation with nausea, diarrhoea and weakness and was found dead in his room 4 days after conservative therapy was begun for presumed cholecystitis. The family did not agree to an autopsy, but interrogation of his implantable defibrillator failed to reveal evidence for an arrhythmia as a cause for his death. Thus, the primary end point, the 30-day major adverse event rate, was 2.8% (1 of 36 patients) for the intention-to-treat population.
There were no fatigue related fractures of the modified Carillon device used in this study, a marked change from TITAN in which a 25% (9/36) incidence of device fracture was seen. The only device fracture in this study was seen in the patient in whom the device could not be recaptured, leaving a recaptured/redeployed, damaged proximal anchor in the middle of the great cardiac vein, where dynamic venous compression was visible (figure 3). The fracture was first seen at the 3-month follow-up, and at the 6-month follow-up a small fragment was seen to be dislodged, without clinical sequelae. The patient did not have any significant clinical events or echocardiographic changes from the procedure over the 12-month follow-up. Fracture due to fatigue of a correctly implanted device, rather than device damage, was not noted.
Secondary end points
The 1-year mortality was 23% (7 of 30 patients). The average time from the procedure to death was 128 days. No deaths were adjudicated to be device related by the Data and Safety Monitoring Board.
Table 2 shows the improvements that were seen in clinical measures.
NYHA class and 6MWT data at baseline to 1, 6 and 12 months
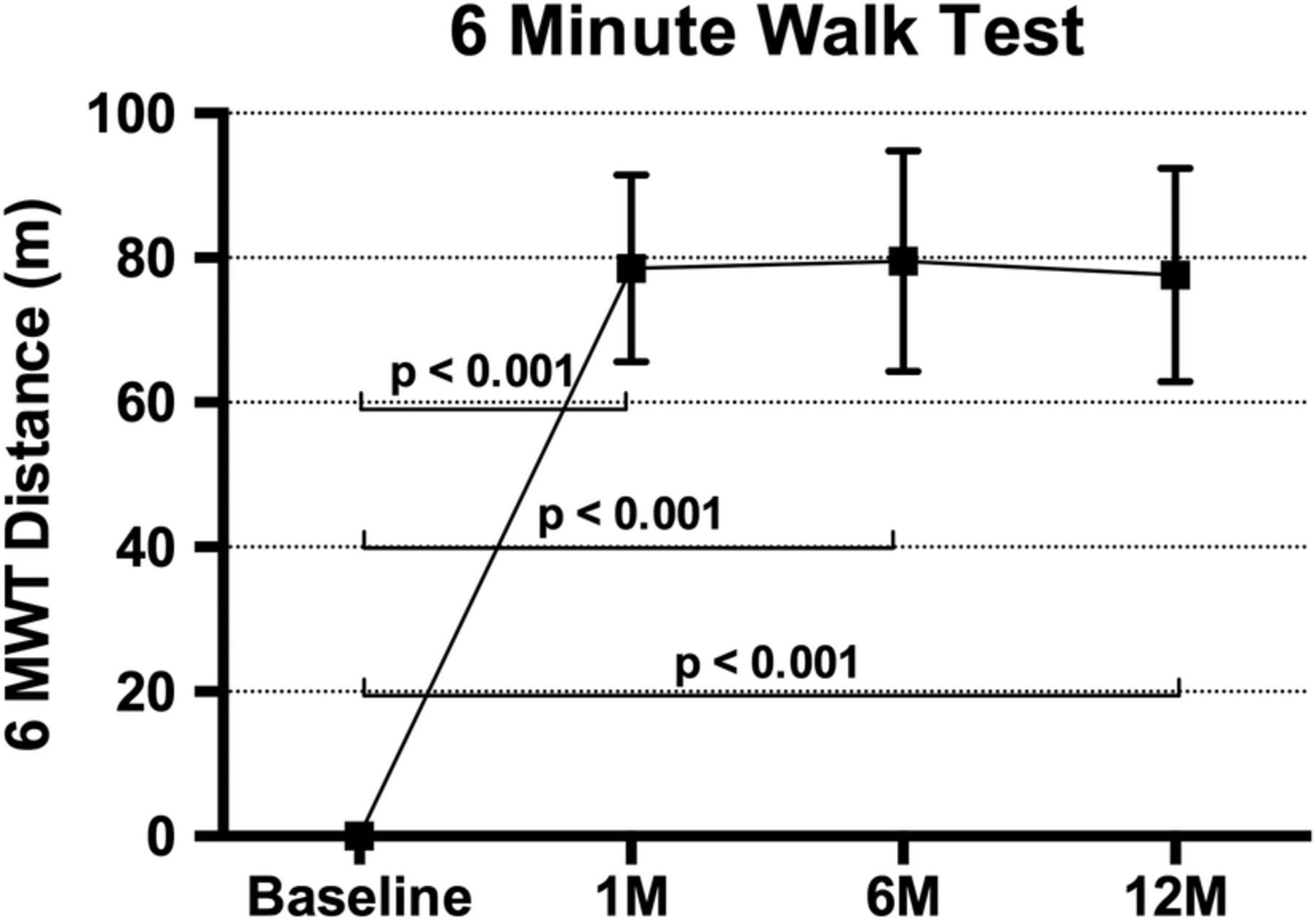
The NYHA grade improved by one grade at 1-month follow-up in 79.3% of patients, and this persisted, being seen in 77.3% at 12 months. Mean improvement in 6MWT at 1 month was 78 m (p<0.01), 80 m at 6 months (p<0.01) and 78 m at 12 months (p<0.01; figure 4).

The 6MWT results showing improvement from baseline at 1, 6 and 12 months in patients receiving an implant (mean±SE of mean). Adjusted for correlation within repeated measures on the same participant and also adjusted for baseline value of the measure (ANCOVA). 6MWT, 6 min walk test; ANCOVA, analysis of covariance.
Improvements were seen in echocardiographic parameters. Echo data were lost in six patients, having been deleted from the local echo workstation before being uploaded to a central storage. Sixty-four per cent of patients had grade 3 to 4+ MR at baseline, compared with only 25% at 1-year follow-up (figure 5). Improvements in quantitative parameters of MR are shown in figure 6, with all parameters showing statistically significant reductions compared with baseline.
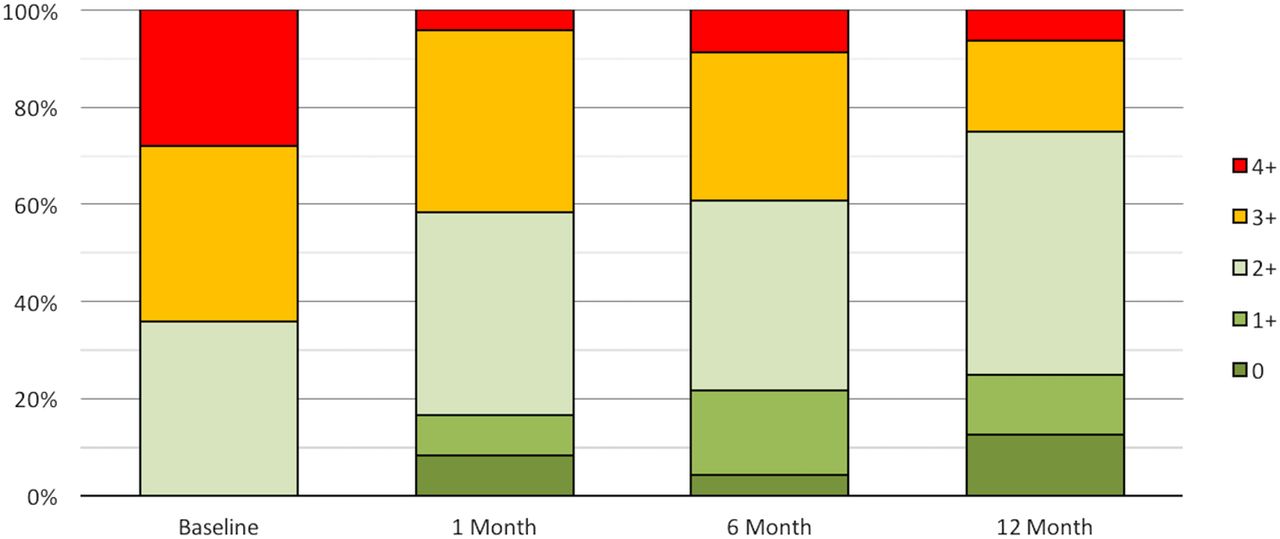
MR severity shown as proportion of patients in each MR grade at baseline, 1, 6 and 12 months. Core laboratory assessed. MR, mitral regurgitation.
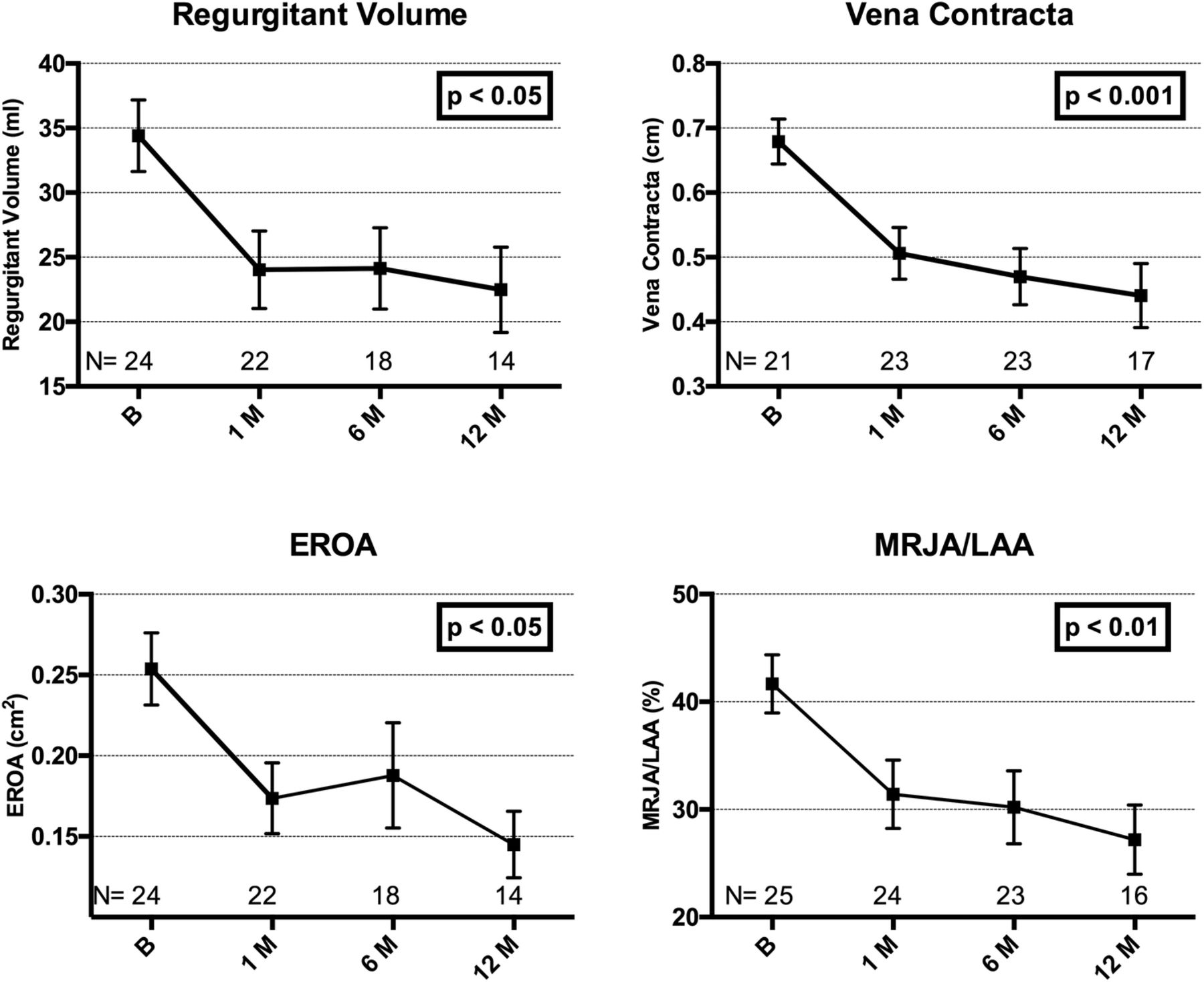
Quantitative parameters of mitral regurgitation demonstrating improvements from baseline (mean±SE of mean). Adjusted for correlation within repeated measures (ANOVA). Core laboratory assessed. ANOVA, analysis of variance; EROA, effective regurgitant orifice area; MRJA/LAA, mitral regurgitation jet area divided by the left atrial area.
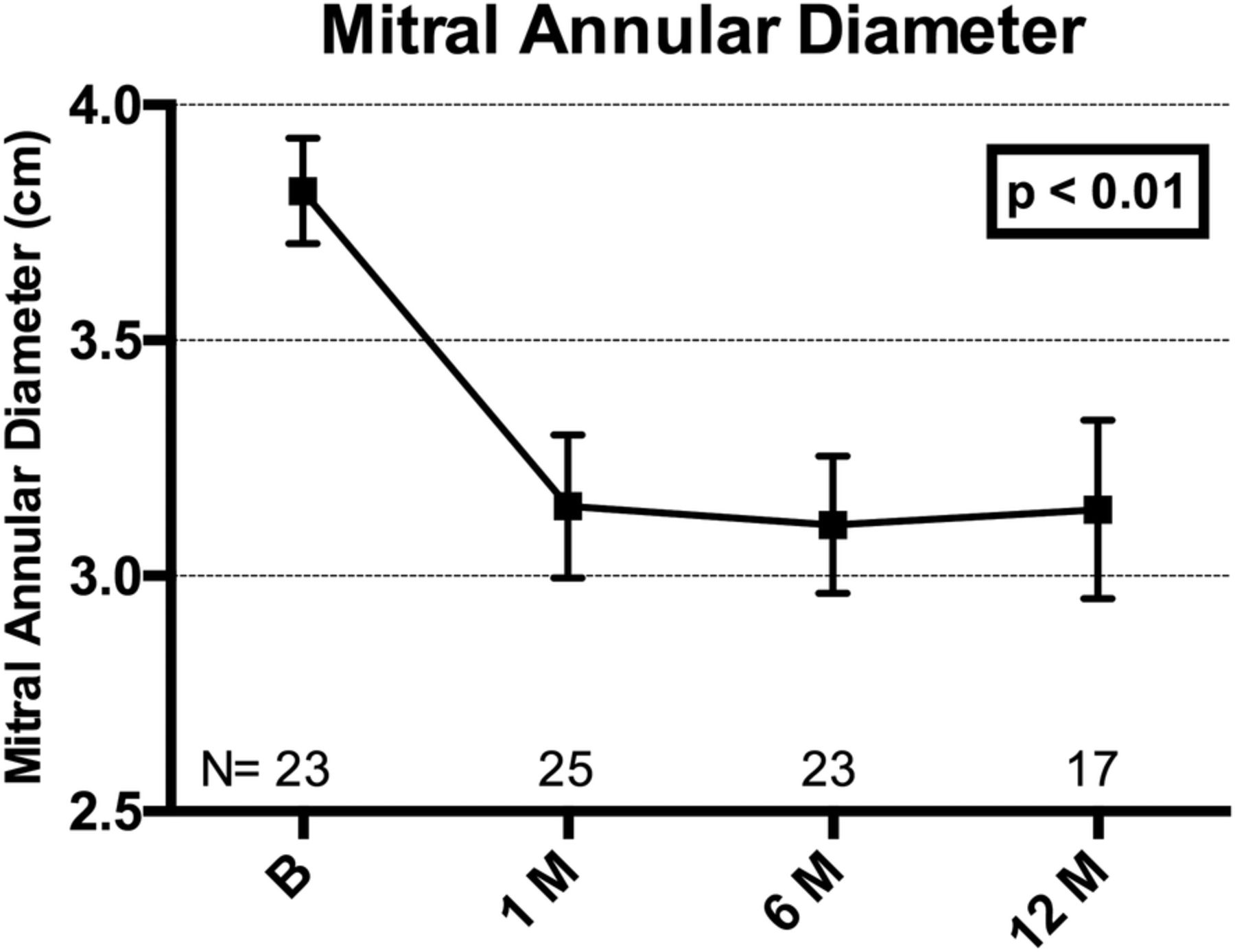
Mitral annular dimensions were reduced with the Carillon device. The anterior–posterior diameter, felt to be the parameter most impacted by FMR,18 demonstrated an ∼15% reduction at all time points over 12 months, p<0.01 (figure 7).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Plot of mitral annular (anterior–posterior) diameter at baseline, 1, 6 and 12 months, showing mean±SE of mean. Adjusted for correlation within repeated measures (ANOVA). Core laboratory assessed. ANOVA, analysis of variance.
Heart failure-related hospitalisation (HFH) data showed that 80% of the implanted population had at least one HFH in the year prior to the Carillon procedure (mean HFH 1.48±1.2). After the Carillon procedure, only 33% of the implanted population had an HFH (mean HFH 0.53±1.0).
Discussion
TITAN II represents the third safety and efficacy study of the Carillon Mitral Contour System. The primary end point of the 30-day rate of major adverse events was similarly low in this study, as it was in AMADEUS and TITAN.15 ,16 This excellent safety profile of the Carillon device and procedure is notable, especially given the high-risk population of symptomatic patients with large left ventricles and low ejection fractions treated in these three studies. As in TITAN, there was only a single major adverse event, a death after hospital discharge, not clearly related to the Carillon procedure. Also, similar to TITAN, there were no device-related clinical complications, and specifically there were no device-related myocardial infarctions.16 The three studies (AMADEUS, TITAN and TITAN II) varied in the use of slightly modified devices in each trial. A significant finding in TITAN II is the lack of fatigue-related fractures of the anchor wireforms, which were seen in 25% of devices in the TITAN study. Although there was one fracture of a proximal anchor seen in TITAN II, this is attributable to the incorrect placement of a recaptured/redeployed (and therefore damaged), proximal anchor in a dynamic portion of the great cardiac vein. This placement was necessitated by the inability to recapture the distal anchor, an unusual occurrence related to distal anchor placement in a small calibre, tortuous vein segment. This scenario can be avoided by attention to this anatomical characteristic when placing a distal anchor.
There were similar clinical and echocardiographic benefits seen in TITAN II as there were in TITAN. This includes improvement in quantitative parameters assessing MR, and reduction in mitral annular dimensions, as well as improvement in NYHA class and 6MWTs. There was also a trend towards a reduction in ventricular size (data not shown), similar to that seen in TITAN. Thus, it appears that the modification of the device used in this study had similar functionality to the device used in TITAN.
The safety and efficacy of the Carillon device shown in this and the prior studies compares favourably to other percutaneous therapies used to treat FMR, with shorter procedure times and comparable (if not superior) periprocedural major adverse event rates and similar clinical benefits.19 ,20
Limitations
The number of patients with adequate quality follow-up imaging for quantitative measurements was low, partially as a result of loss of storage data of echo images involving six patients at one of the most actively enrolling sites. However, the echo findings were consistent with the findings in AMADEUS and TITAN. So even though the ability to draw conclusions from the echo measurements in this study is limited, the consistency between the trials is meaningful. Clinical benefits, such as improvement in the 6MWT and assessment of NYHA class, can be prone to biased assessments. Heart failure hospitalisations were based on site-reporting and were not systematically evaluated. There was no comparable population, with non-implanted patients only followed for safety analysis at 1 month, unlike the TITAN study where non-implanted patients were followed identically to implanted patients over 12 months. Of note, the implant rate was higher in TITAN II than in TITAN, 83% vs 68%. Overall, the findings are consistent with those in the two prior studies with this therapy, and the improvements in the 6MWT continue to be significantly greater than what was historically seen for cardiac resynchronisation therapy.21
Conclusions
The modified Carillon Mitral Contour System used in TITAN II was associated with a low rate of procedure-related complications, and improvements in clinical and echocardiographic measures of MR severity. The outcomes are similar to those seen in the TITAN study, without the fatigue-related fractures seen in the previous iteration of the device, and with a higher implant rate.16 The modified Carillon device used in the TITAN II study is now being assessed in a randomised, blinded clinical trial—the REDUCE FMR trial (NCT02325830).
Acknowledgments
The authors would like to acknowledge the assistance of Nawzer Mehta, PhD, and Ted Feldman, MD, for their review of the manuscript and suggestions.
References
Footnotes
Contributors JL was involved in design of the work, acquisition of data and drafting the manuscript. TS, HS and JM-E were involved in design of the work, acquisition of data and critical revision of the manuscript. HD was involved in design of the work and acquisition of data. JCW was involved in analysis and interpretation of data. CS, PK and IH were involved in acquisition of data and critical revision of the manuscript. DR and SLG were involved in design of the work and drafting the manuscript. MH was involved in design of the work, acquisition of data and critical revision of the manuscript.
Funding This study was funded by Cardiac Dimensions®, Inc.
Competing interests JL is the consultant (proctor) for Cardiac Dimensions, Inc. TS and JCW received research support from Cardiac Dimensions, Inc. HS received consulting and speaker fees from Cardiac Dimensions, Inc. SLG is the consultant (Chief Medical Officer), stock options for Cardiac Dimensions, Inc. MH received consulting (proctor) and speaker fees from Cardiac Dimensions, Inc.
Patient consent Obtained.
Ethics approval Ethics committees in all participating countries.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement No additional data are available.