Article Text

Abstract
Objective To determine predictors of poor outcome in patients with heterotaxy syndrome.
Methods A retrospective review of children with heterotaxy syndrome, in a single tertiary paediatric cardiology centre, was conducted between 1 January 1997 and 1 January 2014 to determine predictors of poor outcome. Poor outcome was defined as death, cardiac transplantation or New York Heart Association (NYHA) functional class III or IV.
Results There were 35 patients diagnosed with heterotaxy syndrome, 17 of whom were diagnosed antenatally. 22 patients had right atrial isomerism and 13 had left atrial isomerism. The median age of postnatal diagnosis was 2.5 days old (1 day to 19 months). 12 patients had a poor outcome; 6 patients died, 1 underwent cardiac transplantation and 5 had an NYHA functional class of >III. 5 patients had a biventricular repair and the remaining 30 had a univentricular repair. Type of atrial isomerism, univentricular or biventricular anatomy, severity of atrioventricular valve regurgitation or ventricular dysfunction, obstructed pulmonary venous return, occurrence of arrhythmia and presence of pulmonary atresia did not predict poor outcome. Fetal diagnosis also did not confer a survival advantage. The median duration of follow-up in this cohort was 65 months (2 days to 16.8 years).
Conclusions Survival for patients with heterotaxy syndrome was 83% over a median follow-up of 65 months. 34% of patients had a poor outcome. None of the variables studied were predictive of death, transplantation or NYHA classification III or IV.
- CONGENITAL HEART DISEASE
- Heterotaxy
- Right atrial isomerism
- Left atrial isomerism
- Survival
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Key questions
What is already known about this subject?
Historically, patients with heterotaxy syndrome had a poor outcome with a high mortality. However, many studies have now documented an increased survival in patients with heterotaxy in more recent years.
What does this study add?
This study reports a high rate of transplant-free survival in one tertiary centre for patients with heterotaxy. No variables studied were found to be predictive for death, transplantation or New York Heart Association class >III. Certain structural defects, such as total anomalous venous drainage and atrioventricular valve regurgitation, were not statistically associated with poor outcome.
How might this impact on clinical practice?
Since the outcome of patients with complex congenital heart disease and heterotaxy is comparable to that of patients without heterotaxy but similar congenital heart defects, this may influence clinicians in how they counsel patients and guardians about active management.
Introduction
Heterotaxy syndrome is defined as an abnormal arrangement of viscera across the left–right axis that differs from complete situs solitus and complete situs inversus.1 ,2 Both variants are associated with complex intracardiac abnormalities. In right atrial isomerism (RAI), compared to left atrial isomerism (LAI), there is a higher incidence of a single ventricle physiology, complete atrioventricular septal defect, total anomalous pulmonary venous drainage (APVD) and pulmonary stenosis.3 The long-term outcome of patients with heterotaxy syndrome is most often dictated by the severity of their cardiac anomalies. Asplenia, which is characteristic of right atrial isomerism, also increases the risk of serious bacterial infection.
Heterotaxy syndrome occurs in ∼0.8% of patients with congenital heart disease.4 Historically, patients with heterotaxy syndrome had a poor outcome with high mortality. RAI has been described by some as one of the worst forms of contemporary heart disease.5 However, many studies have documented increased survival in patients with heterotaxy in recent years. In 1996, a retrospective review of patients with heterotaxy syndrome who underwent Fontan completion at the Mayo Clinic compared outcomes from an early cohort (1973–1986) to those of a later cohort (1987–1992).6 It found that 1-year survival increased significantly from 79% to 88% and 5-year survival increased significantly from 73% to 81%. A more recent retrospective review of outcomes of patients with RAI after surgical management of their cardiac lesions showed that 5-year survival improved significantly in their 2004–2010 cohort compared to their 1997–2003 cohort (81.7% vs 53.8%).7
The aim of this study was to determine predictors of poor outcome in patients with heterotaxy syndrome.
Methods
We undertook a retrospective study of all patients with heterotaxy syndrome presenting to one national cardiac centre over a 17-year period between 1 January 1997 and 1 January 2014. Consent to undertake the study was waived by the Hospital's Ethics Board. Patients with heterotaxy were identified by reviewing echocardiography and catheter laboratory reports while searching for the following terms: heterotaxy, RAI, LAI and interrupted inferior vena cava. The relevant patients' medical, surgical and catheter laboratory notes, laboratory results and imaging studies (including echocardiograms, chest and abdominal X-rays, abdominal ultrasound, cardiac MRI and cardiac angiography) were reviewed by two investigators.
The following data were recorded: current status (deceased, alive or postcardiac transplant), New York Heart Association (NYHA) functional class if alive and without transplant and whether the diagnosis was reached in the antenatal or postnatal period. We assessed the cardiac situs, aortic arch sidedness, anatomy of systemic venous drainage, hepatic venous drainage, APVD (± obstruction), univentricular or biventricular cardiac anatomy, presence of pulmonary atresia, coexistence of cardiomyopathy, atrioventricular valve function and systemic ventricular function. The surgical interventions undertaken were documented. Electrocardiogram and Holter monitor reports were assessed to determine the prevalence and type of arrhythmias. An abdominal ultrasound was undertaken routinely in all patients to identify if a spleen or polysplenia was present. In those with asplenia or polysplenia diagnosed from ultrasound imaging, a blood film was also undertaken to look for Howell-Jolly bodies to ascertain if there was splenic function. Episodes of sepsis were recorded. The presence of ciliary dyskinesia and gut malrotation was considered for review, but the majority of patients were not screened for these conditions. Analysing their relationship to outcome was therefore not possible. Echocardiograms were reviewed by a cardiologist who was blinded to patient identification. Atrioventricular valve dysfunction was classified as mild, moderate or severe and systemic ventricular function as mild, moderate or severe impairment.
Statistical analysis
All statistical analyses were carried out using SAS Statistical Software V.9.4 (SAS Institute, Cary, North Carolina, USA). A binary variable, poor outcome, was defined as a patient who had died or received a cardiac transplant or had an NYHA functional class of III or IV. The variable was set to 1 as a default and set to 0 if none of these conditions were met. Univariate analysis was used to test for independence of the poor outcome variable with 16 other variables of interest. Since the data are binary or categorical, we used the χ2 test. Owing to multiple testing, the significance cut-off was set to 0.003. Those variables exhibiting a p value lower than this cut-off would be used in multivariate analysis using logistic multiple regression. A Kaplan-Meier survival curve for survival and freedom from transplant, comparing RAI and LAI, was also generated.
Results
There were 35 patients with heterotaxy syndrome and congenital cardiac defects identified during the study period. Twenty three (66%) were male. Twenty-two patients (63%) had RAI. Seventeen patients (49%) were diagnosed antenatally. The median age of postnatal diagnosis was 2.5 days (<1 day–19 months). Six patients (17%) died and 1 patient underwent cardiac transplantation. Five of the remaining 28 patients (18%) had an NYHA functional class of III or IV. No patients were lost to follow-up with the median duration of follow-up being 65 months (2 days to 16.8 years). A summary of these findings is seen in table 1.
Demographics, form of atrial isomerism, age at diagnosis and current status
Ventricular and atrioventricular valve function
Systemic ventricular impairment was ≤ mild in 26 patients (74%), moderate in 8 patients (23%) and severe in 1 patient. Atrioventricular valve dysfunction was ≤ mild in 21 patients (60%), moderate in 11 patients (31%) and severe in 3 patients (9%).
Pulmonary venous abnormalities
Fourteen patients had anomalous pulmonary venous drainage; supracardiac in 8 (57%), cardiac in 5 (36%) and infra-diaphragmatic in 1 patient (7%). The pulmonary venous drainage was obstructed in 3 of these patients (21%), of which 2 were supracardiac and 1 cardiac.
The cardiac anatomy for this cohort is summarised in table 2.
The relationship of poor outcome with 16 variables including timing of diagnosis, cardiac anatomy and function, arrhythmias, number of interventions, splenic tissue and sepsis
Surgical outcome
Five patients (14%) reached a biventricular repair, all having LAI. Thirty patients (86%) had a univentricular circulation. Twenty-one patients had a bidirectional Glenn and 10 have gone on to a total cavopulmonary connection. An extracardiac conduit was used in all patients. No patients had an intracardiac lateral tunnel or a classic atriopulmonary Fontan. All but one extracardiac conduit was fenestrated. Six patients (18%) underwent device closure of a fenestration in the catheter laboratory. One patient who completed total cavopulmonary circulation died and another underwent orthotopic cardiac transplantation.
One patient was diagnosed with left ventricular non-compaction cardiomyopathy with a phenotype associated with congenital complete atrioventricular block. The child had severe ventricular dysfunction and valve regurgitation, had a permanent pacemaker implanted and died at age 26 days.
Six patients suffered arrhythmias. Two patients developed supraventricular tachycardia which was medically managed. One patient experienced atrial ectopic tachycardia at 4 months of age and atrial flutter post-surgically. Both were medically managed. One patient developed junctional bradycardia and two had complete heart block. All three patients required insertion of a permanent pacemaker.
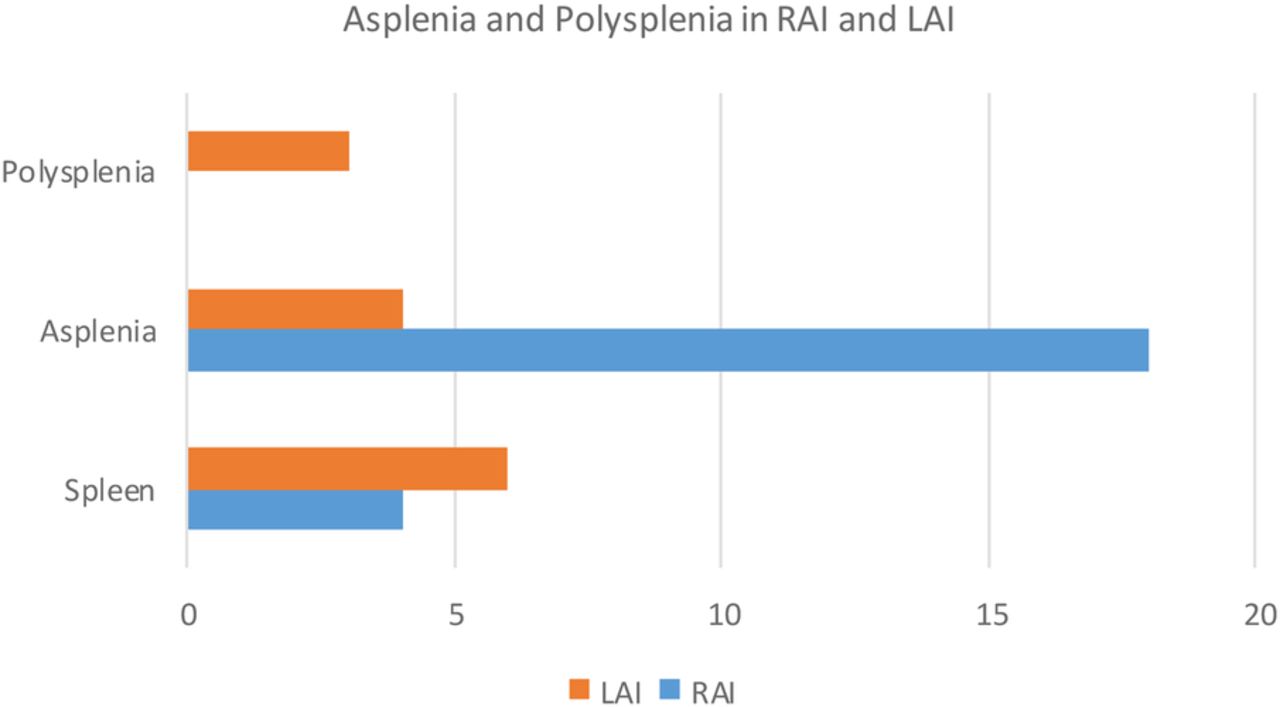
Ten patients (28%) had a normal spleen. Twenty-two patients (63%) were asplenic. Three patients (9%) had polysplenia. Figure 1 outlines splenic tissue found in the study cohort based on atrial isomerism.

Asplenia and polysplenia in right and left atrial isomerism.
Eight patients (23%) had a clinical diagnosis of sepsis, seven of whom also had a causative organism detected. Among the eight patients with sepsis, three were asplenic. Three of the eight patients died, two of whom were asplenic and the other had polysplenia.
Only six children were screened for ciliary dyskinesia with nasal brushings, and they were negative. Five patients were diagnosed with gut malrotation, three of whom had the Ladd procedure. The other two remain unrepaired and asymptomatic.
Predictors of poor outcome
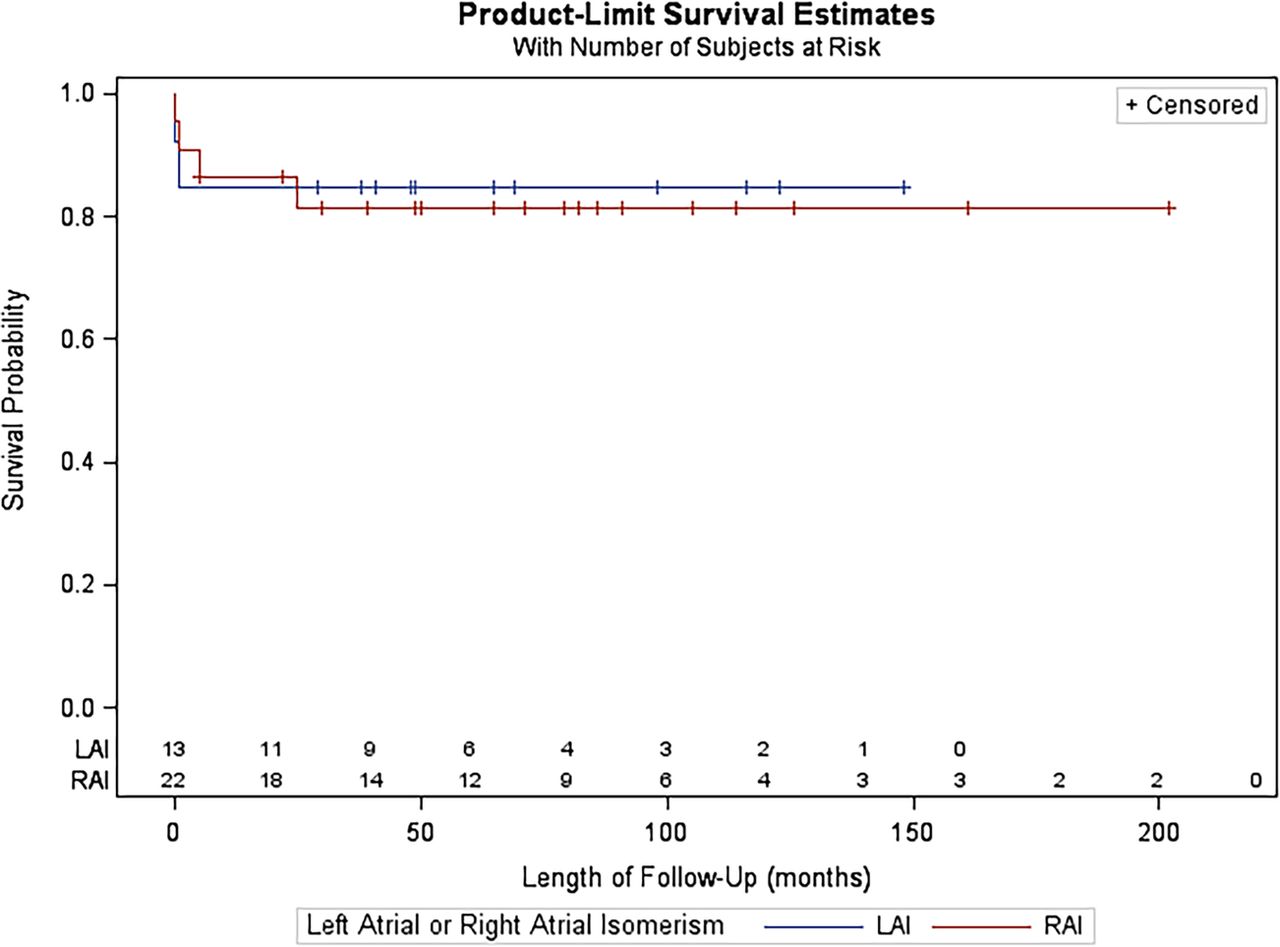
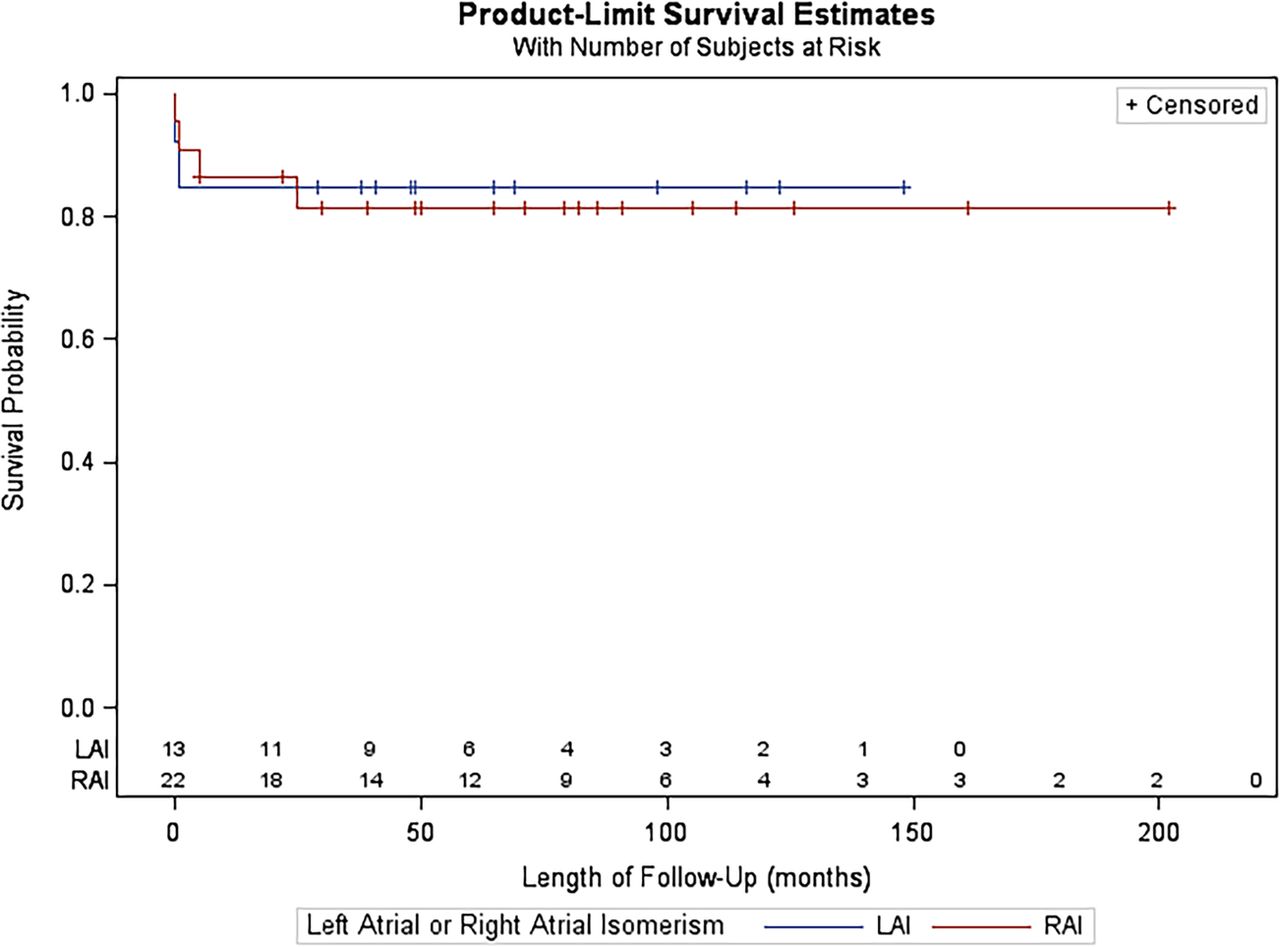
None of the16 variables analysed were significant risk factors for poor outcome, based on univariate analysis (table 2). As a result, multivariate analysis was not carried out. Fetal diagnosis did not confer an advantage for survival for patients in either the RAI or LAI groups. Figure 2 illustrates the Kaplan-Meier plot for freedom from death and transplant comparing patients with RAI and LAI. More patients died or required transplant in the RAI group.
{kind=link}
{kind=link}
Kaplan-Meier survival curves for patients with right atrial isomerism versus left atrial isomerism.
Discussion
The survival of patients with heterotaxy is improving. Our survival rates show figures comparable to other contemporary large centre studies from around the world. Nevertheless, this group of cardiac patients is still felt to be at higher risk than those without heterotaxy syndrome. Patients with complex cardiac lesions and heterotaxy syndrome have frequently been compared to, and do less well than, patients with similar cardiac pathology but no heterotaxy syndrome.8–11 The Society of Thoracic Surgeons' Congenital Heart Surgery Database of North America showed that between 1998 and 2009, the discharge mortality after cardiac surgery for patients with heterotaxy syndrome was higher than that of patients without heterotaxy.8 However, some studies suggest that this discrepancy is starting to narrow. One series showed comparable survival rates between patients with and without heterotaxy who required primary total APVD repair between 1995 and 2005.11 Despite a number of studies now supporting an increasingly comparable long-term survival for patients with heterotaxy, they frequently report increased morbidity in the form of arrhythmias, prolonged pleural effusions and reoperations, especially for atrioventricular valve repair.10 ,12 Lim et al13 described an increased need for intervention in cases where biventricular repair was achieved in patients with heterotaxy. This study reported a low incidence of arrhythmias and a high prevalence of symptom-free survival, with 66% of surviving patients falling into NYHA functional class I or II. However, most of our patients had multiple interventions with 71% having three or more surgical or catheter procedures.
Antenatal diagnosis of complex congenital heart disease and heterotaxy syndrome is increasing. Many studies report antenatal diagnosis of heterotaxy syndrome as achievable, as abnormal viscerocardiac situs can often be identified, along with major intracardiac anomalies and specific markers such as an interrupted inferior vena cava in LAI and juxtaposition of the inferior vena cava and aorta in RAI.14 ,15 Fetal heart block in the first trimester is also indicative of LAI.15 The important question is whether antenatal diagnosis improves patient outcome by reducing morbidity and mortality. One would think that the outcome for antenatally diagnosed patients would be improved, as antenatal diagnosis would facilitate prompt intervention for critical lesions. However, so far studies to demonstrate this have been lacking. Lin et al3 found that half of their patients with RAI died despite antenatal diagnosis and that the mortality rate showed no significant differences when compared to similar postnatally diagnosed patients. Another possible reason why antenatal diagnosis has failed to demonstrate improved outcomes for heterotaxy is that, in LAI, it favourably picks up those fetuses with heart block.14 This group of patients has been described as having a poor outcome as they often suffer fetal demise or early neonatal death secondary to hydrops and heart failure.3 ,15 Escobar-Diaz et al16 found that bradycardia was the only predictor of fetal death in a group of 154 fetuses with heterotaxy from 1995 to 2011. However, the same group also found that the 1-year survival rate of antenatally diagnosed patients with heterotaxy syndrome and atrioventricular block or bradycardia was higher than previously published at 63%.17 Our study failed to show a higher rate of survival for cases that were antenatally diagnosed.
Our analysis did not demonstrate the anatomy of the pulmonary veins or atrioventricular valve dysfunction to be significantly associated with poor outcome. Many large centre studies have demonstrated that the presence of anomalous pulmonary venous connections (especially if obstructed), correctly connected but obstructed/stenosed pulmonary veins, and significant atrioventricular valve regurgitation are risk factors for increased mortality in patients with heterotaxy.9 ,11 ,12 ,18 ,19 Lim et al,13 who studied patients with heterotaxy who had a biventricular repair, found that atrioventricular valve regurgitation and anomalous pulmonary venous connections did not increase mortality in these patients and felt that this should be taken into consideration when contemplating borderline cases for biventricular repair. However, an unbalanced complete atrioventricular septal defect was associated with a higher mortality and pulmonary stenosis, pulmonary atresia and a common atrioventricular valve was significantly associated with increased rates of reoperation.13 Five of our patients (14%) had a biventricular repair and they all have good outcomes (no death or cardiac transplant and NYHA class of I). Two of these patients had a balanced complete atrioventricular septal defect, 1 of whom had severe atrioventricular valve regurgitation requiring surgical repair. The other patient had partial APVD.
There appears to be an emerging consensus among cardiologists and cardiac surgeons that repairing APVD at first operation and repairing a regurgitant atrioventricular valve before the Fontan operation leads to a better outcome.7 ,11 ,18 It is felt that while clinically non-obstructed total APVD remains unrepaired, there may in fact be subtle degrees of obstruction that result in elevated or labile pulmonary vascular resistance.18 It is important to note that in patients with heterotaxy, there is the potential for ongoing occult pulmonary venous obstruction. Many authors have described intrinsically small and abnormal pulmonary veins associated with clinically non-obstructed total APVD, which carry the risk of developing stenosis and increasing risk of death.7 ,20 Our study did not demonstrate that total APVD, with or without obstruction, was statistically associated with poor outcome.
Twenty-two of our patients (63%) were asplenic. Eight patients suffered episodes of sepsis, three of whom were asplenic. Absence of a spleen confers decreased immunity from encapsulated bacteria and increased risk of sepsis. It is very important that asplenic patients receive vaccines against encapsulated bacteria at the earliest possible time and that they comply with lifelong daily penicillin prophylaxis. Interestingly, Chiu et al21 found that patients with heterotaxy syndrome had an increased risk of community-acquired severe bacterial infection and higher mortality compared to other complex cardiac patients, whether a spleen was present or not. Our study did not show absence of a spleen to be associated with poor outcome.
This study has several limitations. It is a retrospective, descriptive study involving 1 centre and only 35 patients. This is a small sample size making statistical analysis of poor outcome challenging. This cohort of patients was not matched to patients with similar cardiac anatomy but without heterotaxy. Any patients who were diagnosed with heterotaxy antenatally and subsequently terminated, or for whom active management was not requested by the parents, were not included in this study.
Survival for patients with heterotaxy syndrome was 83% over a median follow-up of 65 months. Thirty-four per cent of patients had a poor outcome. None of the 16 variables studied were found to be predictors of poor outcome, but this may be a function of the small sample size. Since outcomes for patients with complex congenital heart disease and heterotaxy are comparable to those for patients with similar heart disease and no heterotaxy, this may influence the way in which clinicians counsel patients and their guardians about active management.
References
Footnotes
Competing interests None declared.
Ethics approval Our Lady's Hospital For Sick Children, Crumlin Ethics Committee.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement No additional data are available.