Article Text

Abstract
Objective To present phenotypic characteristics and biomarkers of a family with the rare mutation Thr410Ala of the α-galactosidase A gene (T410A/GLA) causing Fabry disease (FD).
Methods and results In a woman in her 60s with hypertrophic cardiomyopathy, T410A/GLA was found in screening for variants in 59 cardiomyopathy-related genes. Her son in his 40s, two granddaughters and two great grandsons carried T410A/GLA. The son had a history of hypertension and paroxysmal AF but no microalbuminuria or classic symptoms or signs of FD. Baseline α-galactosidase A enzyme (α-Gal A) activity varied from 0% to 26.5%. Cardiac MRI showed mild Fabry cardiomyopathy (FC). During 11 years of enzyme replacement therapy (ERT), FC progressed and he suffered sudden cardiac death in his 50s. The great grandsons with T410A/GLA had no active α-Gal A, high lyso-Gb3 levels and normal cardiac imaging. They suffered from neuropathic pain and gastrointestinal symptoms and were started with ERT at the age under 10. Granddaughters with T410A/GLA had α-Gal A activities of 8–18 and 10% of normal. The older granddaughter in her 30s was diagnosed with incipient FC. Plasma lyso-Gb3 analogues were elevated, markedly in the elder male with FC and moderately in the elder granddaughter. In young males with classic phenotype, plasma lyso-Gb3 analogues were only slightly elevated.
Conclusions The T410A/GLA mutation caused late-onset FD with progressive cardiomyopathy in elder male, and classic FD in young males of the same family. Varying levels of α-Gal A and lyso-Gb3 analogues reflected variable phenotype of FD in the family.
- Cardiomyopathy, Hypertrophic
- Biomarkers
- Metabolic Diseases
Data availability statement
Data are available on reasonable request.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
WHAT IS ALREADY KNOWN ON THIS TOPIC
T410A/GLA is a rare mutation causing classical Fabry disease (FD), with early-onset neuropathic pain and left ventricular hypertrophy.
WHAT THIS STUDY ADDS
Manifestations of FD in the family may vary significantly not only between males and females but also between males with T410A/GLA. Cardiomyopathy may progress during recommended dose enzyme replacement therapy (ERT) and cardiovascular medication even if there are only mild myocardial changes with no fibrosis when ERT is started.
HOW THIS STUDY MIGHT AFFECT RESEARCH, PRACTICE OR POLICY
All family members with the GLA mutation should be carefully monitored by biomarkers and cardiac imaging since childhood and all treatment decisions should be made individually. In addition to the type of the FD causing mutation, and age and gender of the patient, also α-Gal A activity and levels of lyso-Gb3 and its analogues may give a clue to the evolution of the phenotype in the patient. In patients with progressive fabry cardiomyopathy and risk factors for sudden cardiac death, consider implantable cardioverter-defibrillator.
Introduction
Fabry disease (FD) is an X chromosome-linked lysosomal storage disease caused by mutations in the α-galactosidase A gene (GLA). Patients with pathological GLA mutations have decreased levels of functional α-galactosidase A enzyme (α-Gal A) leading to progressive accumulation of glycospingolipids, mainly globotriaosylceramide (Gb3) and globotriaosylsphingosine (lyso-Gb3), in the lysosomes of several organs, interfering with organ function in the long term. Previously, FD was considered rare, affecting only 1 in 40 000–1 in 117 000 live male births. Large-scale newborn screening studies, however, have shown unexpectedly high prevalence of FD causing mutations suggesting incidence of 1 in 3100–4600 individuals.1
Of the over 1000 GLA variants reported, at least 60% are missense mutations.2 Most often, each family has its own rare mutation, limiting evidence for its pathogenicity.3 4 Genotype–phenotype correlations of many GLA mutations are still poorly understood. As a practical approach, FD is divided into classic or late-onset form of FD. Classic form of FD is less common, and it is caused by severe defects of the GLA, such as deletions, or mutations leading to stop codon, but also with severe missense mutations, leading to absent or near absent α-Gal A activity under 5% in hemizygous males. Classic FD is characterised by severe symptoms like neuropathic pain and gastrointestinal problems early in childhood, and progressive kidney failure, brain lesions and cardiomyopathy before middle-age. Angiokeratomas and cornea verticillata may be present from birth. In the more common late-onset FD, missense mutations of the GLA are less severe, and men have residual α-Gal A activity over 5%. Patients remain quite asymptomatic until the third decade of life when cardiomyopathy, the most common and often only manifestation of the disease, and neurological and renal manifestations appear.5 6
The phenotype of FD is affected by not only the gene defect, but also gender and age. In heterozygous women, disease severity varies according to the mutation type and random inactivation of the X chromosome.7 The clinical presentation may also vary in the same family with the same GLA mutation, probably depending on the level α-Gal A activity and other, unknown reasons.3 8 Recent evidence suggests that new biomarkers, particularly plasma and urine lyso-Gb3 and their analogues, may reflect not only the metabolic load but also affect pathogenesis of FD.9–13 Particularly, lyso-Gb3 analogues may play role in the pathogenesis of cardiac hypertrophy, a hallmark of Fabry cardiomyopathy (FC).14 A recent study showed associations between some of these analogues and the Left Ventricular Mass Index and the Mainz Severity Score Index in a large cohort of Fabry patients having a late-onset cardiac variant mutation.15 Patients with another cardiac variant mutation (N215S) also showed increased urinary analogue levels even higher than lyso-Gb3 itself.16 Another study revealed that variations in the GLA correlated with plasma and urine lyso-Gb3 levels.14 16
To our best knowledge, no previous reports show that the same missense mutation causes both classic and late-onset FD in males of the same family. In the present study, we report clinical, imaging and biomarker characteristics of a Finnish family with FD due to a rare GLA missense mutation c.1228A>G p.Thr410Ala (T410A/GLA), where the phenotype differed from classic to late onset in males of different generations, and cardiomyopathy progressed in spite of current enzyme replacement therapy (ERT) in the male with late-onset form of the disease.
Methods
Setting
In 2009, a Finnish index patient with hypertrophic cardiomyopathy (HCM) was genetically tested in London as a part of a European multicentre cross-sectional ACES study.17 Therein, she was found to have T410A/GLA, launching a subsequent family study at the Kuopio University Hospital and University of Eastern Finland. Detailed genetic, biomarker, clinical and cardiac imaging studies were performed in the family members with the T410A/GLA mutation. The T410A/GLA mutation carriers were examined and closely monitored according to the Finnish FD protocol18 at the Kuopio University Hospital. After FD diagnosis, the index patient and her son were also followed at the Turku and Kuopio University Hospitals for 2 years when participating in the Shire TKT-028-REP060-Study,19 in which they received ERT.
Genetic analysis
Genetic analyses of the present study were performed in the Genome Centre of Eastern Finland, Kuopio. In the index patient, the genetic screening from a whole blood sample covered coding regions of the 59 genes related to cardiomyopathy, including GLA. In the son of the index patient, the genetic screening from a whole blood sample covered regions of 62 genes related to cardiomyopathy, including GLA. Cascade screening of T410A/GLA was performed with Sanger sequencing in all available relatives (n=13). In men with T410A/GLA (n=3), Sanger sequencing of the whole coding region of the GLA covered also at least 56 bases (maximum 376 bases) around all exons. Additionally, in male carriers of T410A/GLA, we investigated deep intronic variants of GLA, which according to the literature and genetic databases have been associated with FD.20–24 For details, see online supplemental information.
Supplemental material
In silico structural analysis of mutated α-galactosidase A gene protein
Molecular structure of T410A/GLA mutated protein was obtained using in silico structural analysis as previously described.25
Clinical examination and follow-up
T410A/GLA positive family members (n=6) were examined at the Kuopio University Hospital according to the Finnish FD-protocol.18 For details, see online supplemental information.
Cardiac imaging
All T410A/GLA positive family members (n=6) were examined by cardiac ultrasound according to current guidelines. All of them (n=5), except for the female with Rett’s syndrome, were investigated by cardiac MR imaging (CMR) at least once by using 1.5 T full-body scanner (Magnetom AERA, Siemens Healthcare, Erlangen, Germany). All ventricular wall thicknesses were measured in end-diastole. For details, see online supplemental information.
Alpha-galactosidase A enzyme activity and lyso-Gb3 levels at baseline
Αlpha-galactosidase A enzyme activities were measured in all family members with T410A/GLA (n=6) at least once before any treatment for FD. Lyso-Gb3 levels were investigated regularly in mutation carriers since 2014. For details, see online supplemental information.
Lyso-Gb3 analogues
Tandem mass spectrometry analyses were performed in all three males and the elder granddaughter of the index patient with T410A/GLA at the Université de Sherbrooke to discover lyso-Gb3 analogues in plasma, and in the three males, also in urine. Samples were taken in 2019 when the males, but not the female, were on ERT. For details, see online supplemental information.
Results
Genetic findings in the family
In 2009, the index patient with HCM, participating in the ACES study,17 was found to carry a novel missense mutation T410A/GLA (not found in GnomAD, ClinVar classification P), related to FD. The genetic findings of the index patient were confirmed in the Genome Centre of Eastern Finland in Kuopio by screening for variants in 59 cardiomyopathy-related genes including GLA. T410A/GLA but no other pathogenic or likely pathogenic variants were identified.
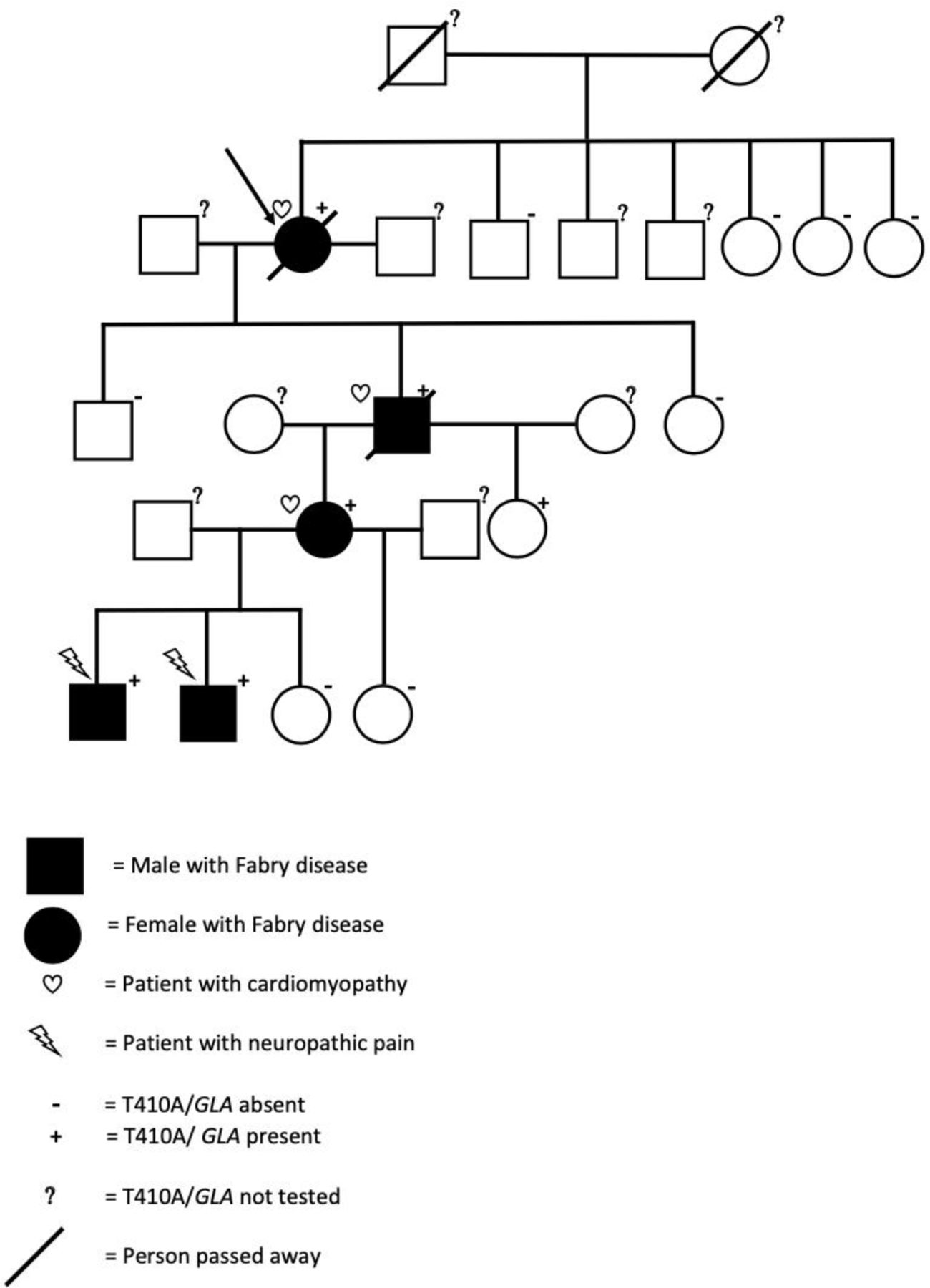
A cascade mutation screening of all available relatives (n=13) was performed with Sanger sequencing (figure 1). In addition to the index patient, shown by arrow in figure 1, five other members of the Finnish family, a son in his 40s, two granddaughters and two great grandsons of the index patient carried T410A/GLA. Three sisters and one brother of index female tested for T410A/GLA did not carry the mutation. The parents of index had passed away and two asymptomatic brothers living abroad were not available for screening.

The family tree of the index patient (arrow) carrying T410A variant of the α-galactosidase A gene (T410A/GLA). Of 13 family members tested, additional 5 were carriers of T410A/GLA and had decreased levels of α-Gal A activity. Cardiomyopathy was diagnosed in the female index patient in her 60s, her son in his 40s and in her granddaughter in her 30s. The two grand grandsons of the index patient had neuropathic pain, gastrointestinal problems and ADHD from early age. ADHD, attention deficit hyperactivity disorder.
In the son with cardiomyopathy, screening for variants in 62 cardiomyopathy-related genes including GLA was performed, and T410A/GLA but no other pathogenic variants were identified.
In the son and two great grandsons carrying T410A/GLA, sequencing all exons of GLA and potentially harmful introns (see online supplemental data) did not reveal any other pathogenic or likely pathogenic variants.
Molecular structure of T410A mutated GLA protein
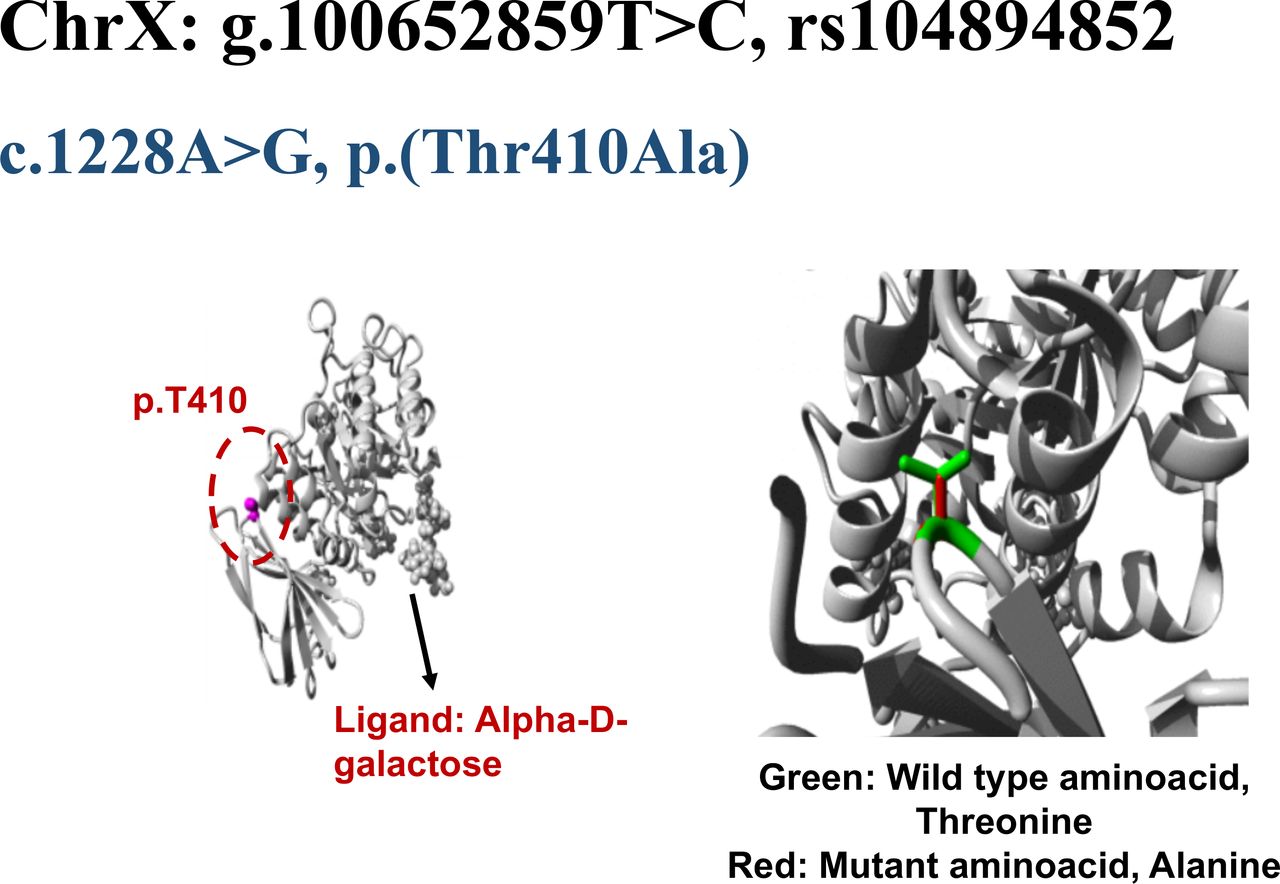
The wild-type residue Threonine (Threonine 410) forms a hydrogen bond with Proline at position 409 (Proline 409; figure 2). The size difference between wild-type and mutant residue not only creates an empty space in protein core but also puts new residue in conformation that does not facilitate a hydrogen bond between Alanine at position 410 and Proline at position 409. The difference in hydrophobicity of wild type Threonine and variant Alanine will also affect hydrogen bond formation. This loss of a hydrogen bond within protein core may disturb correct folding of protein and therefore its function.
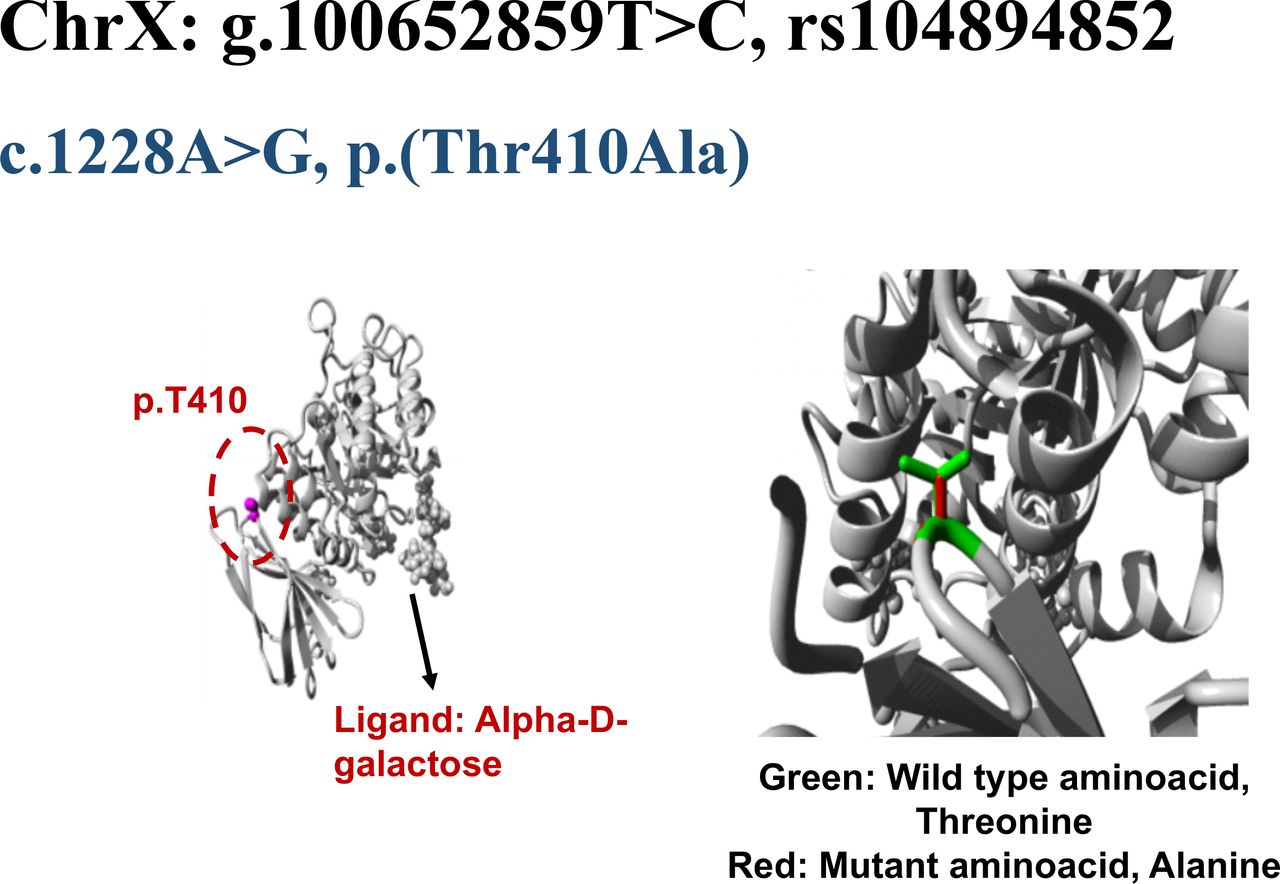
Structural analysis of GLA missense variant, c.1228A>G, p.Thr410Ala. The wild type residue is threonine (Thr410) shown as magenta coloured ball in the core of GLA protein. In the closer view, Thr410 is represented by green side chain whereas the mutant residue, alanine is indicated by red side chain.
History and FD-related clinical and imaging findings of the T410A/GLA mutation carriers
Index patient
In the index patient, α-Gal A activity was 32% of normal. Lyso-Gb3 level measured during ERT was slightly increased (table 1). She had atrial fibrillation (AF), developed dementia and died of stroke in her 70s. (table 1). TnT and proBNP values were slightly elevated at diagnosis. For details, see online supplemental information.
Biomarker and clinical data of the family members with GLA-Thr410Ala mutation
At the time of diagnosis, the echocardiography and CMR of the index female showed moderate left ventricular (LV) hypertrophy with the maximal thickness of 18 mm (figure 3A) in the normal sized (LVESD 31 mm, LVEDI 61 mL/m2 LVESDI 32 mL/m2) heart with normal LV ejection fraction (62%). Mild late gadolinium enhancement (LGE) in the basal inferolateral (BIFL) area, typical of FC, was identified (figure 3B,C). During 7-year follow-up her cardiomyopathy did not progress.

(A–C) The index patient, a female in her 60s. CMR showed signs of mild cardiomyopathy with the left ventricular (LV) maximal thickness of 18 mm in interventricular septum (A, arrow) and mild intramyocardial late gadolinium enhancement in the typical basal inferolateral LV area (B, C, arrows). CMR, cardiac MR.
A male in his 40s
At the time of diagnosis, the son in his 40s had normal kidney function, no microalbuminuria (tables 1 and 2). He had history of hypertension and recent paroxysmal AF but no microalbuminuria or classic symptoms or signs of FD. For details, see online supplemental information.
Fabry biomarkers and CMR findings of the male in his 40s with GLA-Thr140Ala mutation
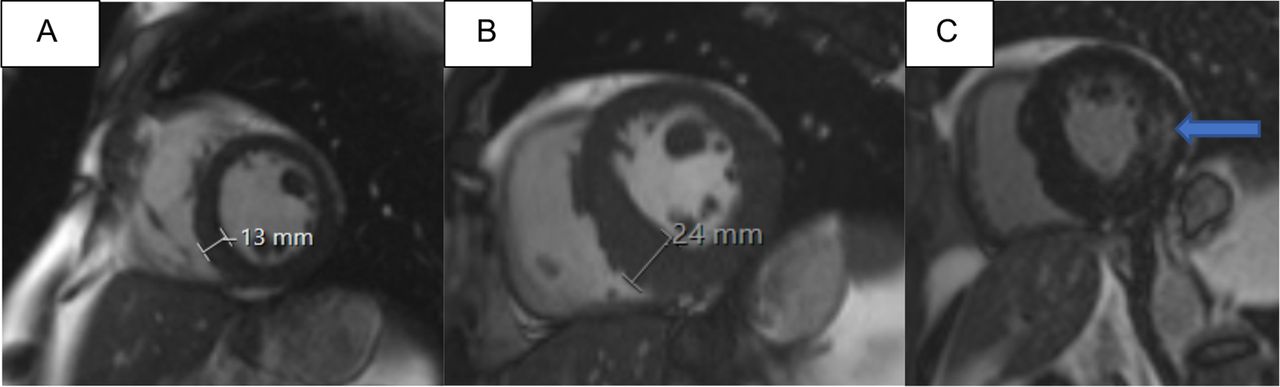
At the time of the diagnosis, there was mild LV hypertrophy (LVH) with maximal LVMWT of 13 mm, normal ejection fraction and no LGE in CMR (table 2, figure 4A). Before starting ERT levels of α-Gal A activity were variable (0 or 2% of normal (table 1)). TnT and proBNP values were normal. CMR was repeated several times during 12 years of monitoring. During the follow-up of 11 years, when he was on optimal ERT (agalsidase alfa 0.2 mg/kg i.v. e.o.w the first 3.5 years and on agalsidase beta 1 mg/kg i.v e.o.w. thereafter) and cardiovascular medication, cardiomyopathy progressed. Finally, CMR showed LVMWT of 24 mm (figure 4B) and moderate myocardial LGE in the BIFL wall of the LV and mild LGE in the basal septum (figure 4C). T1 mapping in recent CMR imaging showed low (<900 ms) values throughout the LV, except for high (>1000 ms) T1 values in the BIFL fibrotic area. His last CMR showed new systolic anterior motion of the mitral valve, the LV mass was 155 g/m2 (normal range 70–113 g/m2), but aortic dimensions had remained unchanged and he had no LV aneurysm. Half a year later, he suffered autopsy verified sudden cardiac death (SCD) in his 50s. The myocardium was thickened and fibrotic, there were signs of lung congestion due to heart failure, but no coronary artery disease, aortic rupture or pulmonary embolism was detected. His sudden death was most likely triggered by a ventricular arrhythmia.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
(A–C) Cardiomyopathy of the male in his 40s with T410A/GLA progressed when he was on enzyme replacement therapy (ERT). At baseline, CMR derived left ventricular (LV) maximal thickness was 13 mm (A) and no late gadolinium enhancement (LGE) was seen at baseline, but 11 years later LV thickness was 24 mm (B) and intramyocardial LGE was seen in the typical basal inferolateral area (C, arrow). CMR, cardiac MR.
A female in her 20s
The elder granddaughter of the index patient had been suffering from abnormal fatigue for years, but had no other symptoms or signs of FD (table 1). In her 30s, CMR showed signs of incipient FC.23 For details, see table 1 and online supplemental information.
A female in her 10s
The younger granddaughter of the index patient was diagnosed with T410A/GLA in her 10s. Because of Rett′s syndrome, she is unable to speak and must be fully assisted in all her daily activities. She has not had symptoms or signs of FD so far. Her cardiac ultrasound, ECG and creatinine, which were last examined in her 20s, were normal but lyso-Gb3 level was slightly elevated.
Two young males
The great grandsons of the index patient have been suffering from neuropathic pain, fatigue and the younger one from gastrointestinal symptoms from early preschool age and they both have been diagnosed with attention deficit hyperactivity disorder (table 1). Their well-being has improved since they were started with ERT under age 10. ECG, cardiac ultrasound and recent CMR findings as well as creatinine and eGFR values have been normal and there has been no microalbuminuria in repeated measurements.
All family members
Angiokeratoma and cornea verticillata, typical findings of classic FD, were not detected in this family.
Alpha-galactosidase A activity and lyso-Gb3 levels
The FD-related biomarkers of the family members with the T410A/GLA mutation are summarised in table 1. All family members with T410A/GLA (n=6) had low or immeasurable α-Gal A activities compatible with FD. The females (n=3) had some own α-Gal A activity varying from 8 to 32% of normal. α-Gal A activity of the son of the index was measured four times and varied from 0% to 26.5%. His last measurement (14% from DBS sample) was performed recently, when he had been on ERT for years. The two great grandsons had no active α-Gal A.
Lyso-Gb3 levels were elevated in all six mutation carriers. Lyso-Gb3 levels of the females were slightly elevated. In the index patient, measurement was performed only once when she was already on ERT (tables 1 and 2). The son had elevated lyso-Gb3 levels up to 13.1 ng/mL during the ERT. Both great grandsons had elevated lyso-Gb3 levels up to 20–27 ng/mL before starting ERT.
Tandem mass spectrometry for plasma and urine lyso-Gb3 analogues
Profiles of plasma lyso-Gb3 analogues showed high levels of total lyso-Gb3 and all but one its analogues in the male carrier with FC (tables 3 and 4). Also in heterozygous female, levels of total and all but one of the analogues were increased, but to lesser extent than in the elder male. In contrast, both young males with classic phenotype had plasma profiles with only slight or nondetectable changes in analogue levels. In the elder male, urine levels of total lyso-Gb3 and all but two of its analogues were abnormally high. In young males, levels of all but one analogues were increased with a pattern of not very different from that of the elder male.
Lyso-Gb3 and analogue levels in plasma of the family members with GLA-Thr410Ala mutation
Lyso-Gb3 and analogue levels in urine of the males with GLA-Thr410Ala mutation
Discussion
Principal findings
In the present family study, we showed that T410A/GLA causes classic FD in two young males and late-onset FD with cardiomyopathy in a middle-aged male, which, to our knowledge, is a novel finding. The phenotype in males was related to α-Gal A activity, lyso-Gb3 levels and profile of lyso-Gb3 analogues, implying the importance of these biomarkers in phenotypic diversity of FD.
In addition, in the middle-aged male with residual enzyme activity, FC progressed during normal-dose ERT and other recommended medications for FC, and the patient suffered SCD due to cardiomyopathy.
In the context of the current literature
T410/GLA variant
T410/GLA variant is a rare mutation not recorded on the gnomAD database, but has been classified as pathogenic on the ClinVar database, and is classified amenable mutation for migalastat treatment (www.galafoldamenabilitytable.com). There is only two earlier publications on the variant found in PubMed, one reporting a Chinese family with five T410A/GLA carriers,26 including three hemizygous males all having classical type of FD with neuropathic pain from their teens. The male in his 30s had also LVH, which was not present in two adolescent males. The α-Gal A activities in the males varied from 7% to 17% of normal value. Two females carrying T410A/GLA had normal α-Gal A activities and no symptoms of FD. On FD screening in Taiwanese cohort of patients with chronic kidney disease, T410A/GLA was found in two males with classic phenotype.27
In the present study, the molecular structure analysis of the T410A mutated GLA protein suggested disturbed folding, futher supporting pathogenicity of T410A/GLA. T410A/GLA was found in the index patient, but not in her four siblings, suggesting that the mutation has arisen de novo, which is common in FD.6 In all six mutation carriers, T410A/GLA caused zero to low enzyme activity levels, and clinical FD in 5 of 6. The two young males with no active α-Gal A had classic FD symptoms starting from age under 10. In contrast, their grandfather with residual α-Gal A activity had typical late-onset FC characterised by cardiomyopathy without history of any symptoms of FD in childhood or adolescence. Females with residual α-Gal A activity had typical late-onset FD with cardiomyopathy. To our knowledge, there are no previous reports showing that the same missense mutation causes both late-onset and classic form of FD in male carriers of the same family.
SCD in FD
According to a systematic review on 13 studies with over 4000 FD patients, 75% of deaths during the 1.2–10 years of follow-up were due to cardiovascular causes, of which 62% were SCDs. The risk factors associated with SCD were age (over 40 years in males), male gender, LVH, LGE in CMR and non-sustained ventricular tachycardia.28 So far, there are no clear models for risk prediction in FC. Using the HCM Risk-SCD Calculator by the ESC is not recommended in FC.29 In the present study, the male with progressive FC and SCD had several risk factors, and transient symptoms of heart failure.
Possible mechanisms for variable phenotype in males carrying T410A/GLA
Additional genetic factors
To exclude other gene variants contributing to FD phenotype in the family, we performed comprehensive genetic studies, including comprehensive cardiomyopathy panels and sequencing of whole coding region and deep intronic variants of GLA. The only pathogenic variant causing FD and cardiomyopathy in this Finnish family was T410A/GLA.
Alpha-galactosidase A enzyme activity
Family members with T410A/GLA had low or immeasurable α-Gal A activities, compatible with FD. α-Gal A activity in the young males was 0%, and they had classic FD. The middle-aged son of the index patient had variable α-Gal A activity in several measurements and late-onset disease. α-Gal A activity may have varied depending on the different methods used, technical variability in enzyme assays, enzyme instability or metabolism changes. Phenotypic presentation may vary in family members depending on α-Gal A activity.
Levels of lyso-Gb3 and its analogues
Levels of plasma lyso-Gb3 analogues were markedly elevated in the elder male with FC and in the heterozygous female with incipient FC. In contrast, in both young males with classic phenotype but no FC, plasma lyso-Gb3 analogues were only slightly elevated. Consequently, as suggested by previous studies lyso-Gb3 analogues may reflect and even modify the phenotype, including cardiomyopathy, in FD.14–16
Clinical implications
First, measuring α-Gal A activity, lyso-Gb3 and possibly also its analogues is important in patients with FD, as phenotypic presentation and progression of the disease may vary depending on the level of biomarkers. We recommend to investigate a complete biomarker profile of lyso-Gb3 and its analogues. Second, regular CMR at least every 2–3 years30 is important in adult subjects with FD irrespective of ERT to recognise progression of FC. Third, in FD patients with progressive LVH and risk factors for SCD, consideration of implantable cardioverter-defibrillator is worthwhile. Finally, in the future, systematic studies to develop SCD risk calculator for FC are needed.
Future perspectives
Future studies are required to investigate the factors influencing α-Gal A activity, lyso-Gb3 levels and its analogues in vivo in carriers of FD causing mutations, and the relation of these biomarkers with clinical phenotype and progression of the disease.
Conclusions
The missense mutation T410A/GLA causes late-onset FD with cardiomyopathy not only in males but also in females, and classic FD in other males, probably depending on α-Gal A activity and levels of lyso-Gb3 and its analogues.
Data availability statement
Data are available on reasonable request.
Ethics statements
Patient consent for publication
Ethics approval
This study involves human participants and was approved by the Research Ethics Committee of the Northern Savo Hospital District414/2016 Fabryn tauti Suomessa (FinFabry). Participants gave informed consent to participate in the study before taking part.
Acknowledgments
The authors would like to thank all participating pediatricians and Professor Markku Laakso for their co-operation.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors KV: study, analysis, reporting, clinical examination, adult echo, writing the manuscript. MH: imaging analyses, revision of the manuscript. IK: study conception, revision of the manuscript. SW: patient data collection, revision of the manuscript. SH, MM and JR: genetic analyses, revision of the manuscript. CA-B: analysis of lyso-Gb3 and its analogues, writing and revision of the manuscript. JK: study conception, analysis, adult echo, writing and revision of the manuscript, guarantor.
Funding The study was supported by Sanofi, the Academy of Finland, the Finnish Heart Research Foundation and the Kuopio University Hospital (grants to JK)
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.