Article Text

Abstract
Objective Anthracyclines are successfully used in cancer treatment, but their use is limited by their cardiotoxic side effects. Several risk factors for anthracycline-associated cardiomyopathy (AACM) are known, yet the occurrence of AACM in the absence of these known risk factors suggests that other factors must play a role. The purpose of this study was to evaluate whether a genetic predisposition for dilated cardiomyopathy (DCM) could be a potential risk factor for AACM.
Methods A hospital-based registry of 162 DCM families and two hospital-based registries of patients with cancer treated with systemic cancer therapy (n>6000) were reviewed focusing on AACM. Selected patients with AACM/DCM families with possible AACM (n=21) were analysed for mutations in cardiomyopathy-associated genes and presymptomatic cardiological evaluation of first-degree relatives was performed.
Results We identified five DCM families with AACM and one patient with AACM with a family member with a possible early sign of mild DCM. Pathogenic MYH7 mutations were identified in two of these six families. The MYH7 c.1633G>A (p.Asp545Asn) and c.2863G>A (p.Asp955Asn) mutations (one double mutant allele) were identified in a DCM family with AACM. The MYH7 c.4125T>A (p.Tyr1375X) mutation was identified in one patient with AACM.
Conclusions This study further extends the hypothesis that a genetic predisposition to DCM could be a potential risk factor for AACM.
- GENETICS
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Key messages
-
What is already known about this subject?
-
Anthracyclines can cause anthracycline-associated cardiomyopathy (AACM). Several risk factors for AACM are known. However, other risk factors must exist as AACM also occurs in the absence of these risk factors.
-
How might this impact on clinical practice?
-
The results of our study provide further support for the hypothesis that a genetic predisposition for familial dilated cardiomyopathy (DCM) could be a potential risk factor for anthracycline-cassociated cardiomyopathy (AACM). Although more research is needed, our preliminary results should alert clinicians to the possibility of AACM if a patient reports a personal and/or familial history of DCM/heart failure. Conversely, if a patient with cancer develops disproportional LV dysfunction after anthracycline treatment, one could consider offering the index patient DNA analysis of cardiomyopathy-associated genes and presymptomatic cardiological evaluation to first-degree family members.
-
What does this study add?
-
In patients with cancer with a family history of dilated cardiomyopathy (DCM)/heart failure, one should be vigilant for increased susceptibility to anthracycline-associated cardiomyopathy (AACM) and one may consider more intensive cardiovascular monitoring before and during cancer treatment or alternative non-cardiotoxic cancer therapy.
-
Identifying a cardiomyopathy-related mutation facilitates genetic cascade screening in family members, which can be helpful in identifying individuals at risk (and in dismissing relatives who do not carry the mutation from regular cardiac monitoring). This enables timely diagnosis to be made, with the possibility of preventing complications and reducing morbidity and mortality.
Introduction
Anthracyclines, such as doxorubicin, epirubicin and daunorubicin, are important drugs in the treatment of cancer. Their use in clinical practice is hampered by their cardiotoxic side effects.1–3 These effects are variable and range from transient electrophysiological abnormalities to anthracycline-associated cardiomyopathy (AACM) and heart failure.1–4 AACM typically presents as left ventricular (LV) dysfunction/dilated cardiomyopathy (DCM) in adults. In children, AACM may also present as restrictive cardiomyopathy.4 ,5 AACM may become apparent during anthracycline therapy, or within the first year thereafter, or later (sometimes even 10–20 years after treatment).4 ,6 It has been suggested that anthracyclines cause cardiac dysfunction through myocyte damage due to excess production of reactive oxygen species.7 ,8 However, other mechanisms, such as anthracycline-dependent inhibition of topoisomerase II β and selective inhibition of cardiomyocyte-specific gene expression, have also been proposed.7 ,9 The risk of AACM depends on the cumulative dose of administered anthracyclines.1 ,2 In adults, the recommended maximum cumulative doses of doxorubicin and epirubicin are (in general) 550 and 900 mg/m2, respectively.10 In children, the maximum cumulative dose of anthracyclines is (in general) 450 mg/m2.11 Additional risk factors for AACM have been identified, including both a younger age as well as an older age at treatment, female gender, mediastinal radiation and pre-existing heart disease.1 ,2 ,12 Yet, the occurrence of AACM in the absence of these known risk factors suggests that other predisposing factors must play a role.
Recently, we reported two patients with AACM with multiple family members diagnosed with DCM, suggesting the presence of a genetic predisposition for DCM in these families.13 In one of these families, we confirmed this finding by identifying a double mutant MYH7 allele. Based on these observations we hypothesise that a genetic/familial predisposition for DCM might be a potential risk factor for AACM.
To further corroborate the concept that a genetic/familial predisposition for DCM might be a potential risk factor for AACM, we have searched for patients with AACM in our registry of DCM families and, reciprocally, searched for the presence of familial cardiomyopathy in patients with AACM among two registries of patients with cancer treated with systemic therapy.
Methods
Patients and clinical evaluation
Patients with AACM were selected from our hospital-based registry of proven DCM families (cohort I14) and from two hospital-based registries of patients with cancer (adult or childhood onset) treated with systemic therapy (cohort II—adult onset patients, cohort III—childhood onset patients15). For the flow chart of patient inclusion, see figure 1. All participants seen at our cardiogenetics outpatient clinic underwent a counselling procedure and agreed to take part in our study. The institutional review committee approved the protocol.

Flow chart patient inclusion. Patients with AACM data were collected from the hospital-based registry of DCM families (cohort I) and two hospital-based registries of patients with cancer (adult- or childhood-onset) treated with systemic cancer therapy (cohort II-adult-onset patients and cohort III-childhood-onset patients). AACM, anthracycline-associated cardiomyopathy; DCM, dilated cardiomyopathy; FU, follow-up; LVEF, left ventricular ejection fraction; SF, shortening fraction; WMSI, wall motion score index.* Two patients from a previously published study.13 †Using the cut-off values of ≤450 mg/m2 for doxorubicin and ≤600 mg/m2 for epirubicin, we aimed to include patients who received at most the submaximal dose of anthracyclines.
Cohort I: Hospital-based registry of DCM families
Patients with idiopathic DCM (and other putative inherited cardiac disorders) are routinely evaluated at the cardiogenetics outpatient clinic of the University Medical Center Groningen, including registration of the patients’ clinical and family history, construction of pedigrees and if indicated DNA analysis. Family members at risk are invited for presymptomatic cardiac screening and/or, in those families where a mutation is identified, presymptomatic DNA analysis. To inform family members at risk, the index patients are asked to distribute letters to their family.16 DCM is diagnosed using the generally accepted criteria by Mestroni et al, that is, reduced systolic function (left ventricular ejection fraction (LVEF) <0.45) and dilation of the left ventricle (LV end-diastolic dimension >117% of the predicted value corrected for body surface area and age) without identifiable causes such as severe hypertension, coronary artery disease and systemic disease.17 Mild DCM is diagnosed if only one of these criteria is fulfilled. Familial DCM is defined by at least two family members with (mild) DCM or by a patient with DCM with a first-degree relative who died suddenly before the age of 35 years. We reviewed all familial DCM index patients (n=162) and their family members registered at the cardiogenetics outpatient clinic as of 1 January 2012 for the presence of AACM. AACM was defined by patients who developed DCM (without coronary artery disease or other identifiable causes) after treatment with cumulative doses of doxorubicin ≤450 mg/m2 and cumulative doses of epirubicin ≤600 mg/m2 (with or without additional trastuzumab therapy). By using cut-off values that are lower than the generally accepted safety margins of <550 mg/m2 for doxorubicin and <900 mg/m2 for epirubicin,10 we aimed to include patients who received at most a submaximal dose of anthracyclines.
Cohort II: Hospital-based registry of adult-onset patients with cancer treated with systemic cancer therapy
Since the late 1980s, the Department of Medical Oncology of the University Medical Center Groningen has routinely collected clinical data on patients with cancer who have been treated with systemic therapy at the University Medical Center Groningen. In this registry, we reviewed all patients (n=6107) for the presence of AACM. AACM was defined by patients diagnosed with a reduced systolic function (LVEF <0.50) after anthracycline treatment, according to the aforementioned cut-off values and after excluding other identifiable causes for LV dysfunction. All patients with AACM who were alive, with no evidence of active malignant disease and with a follow-up of at least 3 years, were informed about the study and asked to complete a questionnaire about their family medical history. Selected patients with AACM who agreed to participate in the study were counselled at our cardiogenetics outpatient clinic (see figure 1 for inclusion criteria). A family history suggestive of DCM was assigned if patients reported one or more relatives with possible heart failure, cardiomyopathy, pacemaker/implantable cardioverter defibrillator (ICD) therapy and/or sudden cardiac death before the age of 60 years (excluding relatives with identifiable causes for cardiac disease or sudden death, such as coronary artery disease).
Patients with an LVEF <0.30 at follow-up, or with a persistent LVEF ≤0.45 and a first-degree relative with possible cardiomyopathy or myopathy, were offered next generation DNA sequencing targeting 48 cardiomyopathy-associated genes and cardiological examination of first-degree family members. All other patients were offered limited DNA analysis of four DCM-related genes.
Cohort III: Hospital-based registry of childhood-onset patients with cancer treated with systemic cancer therapy
Between 1976 and 1999, the Division of Pediatric Oncology, Beatrix Children's Hospital, University Medical Center Groningen has collected the clinical data of childhood-onset patients with cancer treated with systemic cancer therapy who survived at least 5 years after diagnosis. As part of a research project on late effects on cardiovascular damage and risk factors, cardiac examinations (including echocardiography) were performed on 277 patients.15 Using this cohort of 277 patients, we searched for patients with AACM who were treated with a total cumulative dose of doxorubin ≤450 mg/m2 and had (1) a wall motion score index (WMSI) ≥1.5 and a shortening fraction (SF) <0.29, or (2) a WMSI ≥1.5 or an SF <0.29 and a family history suggestive for DCM. All included patients had, apart from their treatment, no other identifiable causes for LV dysfunction. Patients who showed recovery of LV function without therapy during follow-up (LVEF >0.55) were excluded. A family history suggestive of DCM was assigned according to the aforementioned criteria (see cohort II). Patients eligible for our study were counselled at our cardiogenetics outpatient clinic and, upon patients’ consent, next-generation DNA sequencing targeting 48 cardiomyopathy-associated genes was performed.
Genetic analysis
Blood-derived genomic DNA was obtained from all index patients of DCM families with apatient with AACM (cohort I) and the selected patients with AACM from cohorts II and III. All index patients included in cohort I, cohort III, and all patients with an LVEF <0.30 at follow-up or with a persistent LVEF ≤0.45 and a first-degree relative with possible cardiomyopathy or myopathy from cohort II were screened for mutations in 48 cardiomyopathy-associated genes (ABCC9, ACTC1, ACTN2, ANKRD1, BAG3, CALR3, CRYAB, CSRP3/MLP, DES, DMD, DSC2, DSG2, DSP, EMD, GLA, JPH2, JUP, LAMA4, LAMP2, LMNA, MYBPC3, MYH6, MYH7, MYL2, MYL3, MYPN, MYOZ1, MYOZ2, PKP2, PLN, PRKAG2, PSEN1, PSEN2, RBM20, RYR2, SCN5A, SGCD, TAZ, TBX20, TCAP, TMEM4, TNNC1, TNNI3, TNNT2, TPM1, TTN, VCL and ZASP/LDB3) using targeted next-generation DNA sequencing.18 All novel variations were confirmed by Sanger sequencing. In the remaining index patients from cohort II (n=10), we only analysed the complete coding sequence and intron/exon boundaries of four DCM-related genes (PLN, MYH7, LMNA and TNNT2) with Sanger sequencing.19 Details of these analyses are available on request. Classification of the variants identified was performed according to criteria previously published in van Spaendonck-Zwarts et al.19 Variants were classified as not pathogenic, variant of unknown clinical significance (VUS; VUS1, unlikely to be pathogenic; VUS2, uncertain; VUS3, likely to be pathogenic), or (putative) pathogenic. Co-segregation analysis of VUSs and mutations was performed where possible.
Results
Patient selection and cardiological evaluation
Cohort I: Hospital-based registry of DCM families
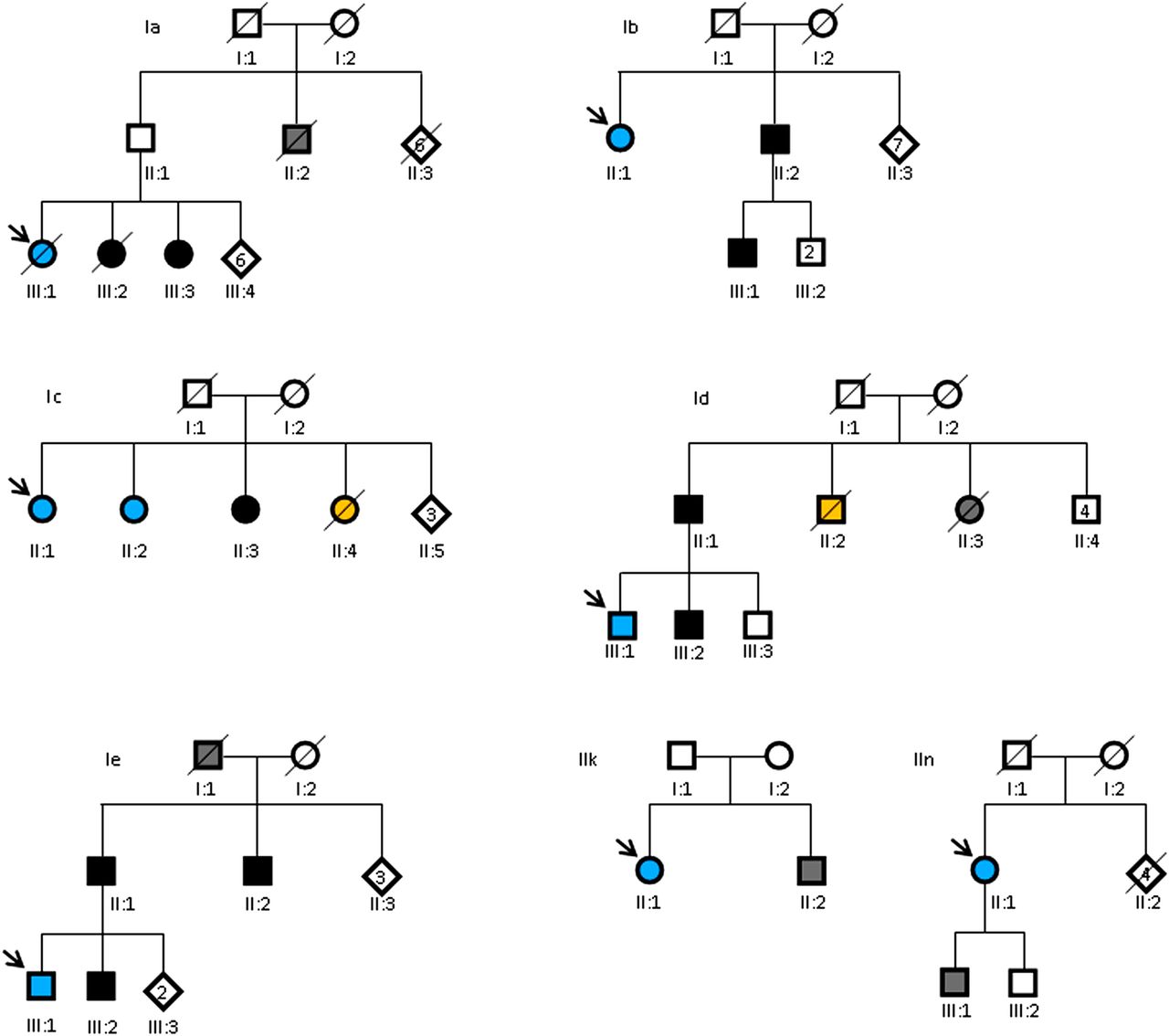
Among our hospital-based registry of 162 DCM families, we identified five DCM families with at least one patient with AACM (figure 1). Two of the five families (Id and Ie) had also been identified in the childhood cancer registry (cohort III) and were previously published.13 In families Ia and Ib, we identified one patient with AACM and two family members with (mild) DCM. In family Ic, we identified three sisters, two of whom had AACM and one DCM. A fourth sister had died suddenly at the age of 62 years. In family Id, we identified two first-degree relatives (father and brother) of the patient with AACM with (mild) DCM. In family Ie, three relatives (father, brother and paternal uncle) of the patient with AACM had (mild) DCM (table 1 and figure 2).
Characteristics of patients with AACM and their family members
{kind=link}
{kind=link}
Pedigrees of patients with AACM and their family members. Square symbols, men; circles, women; diamonds, unknown sex. Blue symbols, patients with AACM; solid black symbols, patients with (mild) DCM; grey symbols, possible DCM; orange symbols, sudden cardiac death. Diagonal lines through symbols, deceased; arrow, index patient; number in a symbol, number of individuals with this symbol.
Cohort II: Hospital-based registry of adult-onset patients with cancer treated with systemic cancer therapy
After applying our selection criteria (figure 1), 43 patients were informed about our study and asked to fill out a questionnaire including questions about family history. Thirteen of these patients were eligible for our study (table 1 (patients IIa through IIn)). Presymptomatic cardiological screening of first-degree relatives was offered to 3/13 patients: patients IIk, IIm, IIn (table 1). Three daughters of the index patient in family IIk underwent cardiological examination. Two showed no signs of DCM, while one was diagnosed with subnormal LV function (LVEF 0.50). Analysis of the pedigree showed that the brother of patient IIk was diagnosed with a dilated LV with preserved function and atrial fibrillation (AF) (see figure 2). Family members of patient IIm did not respond to our invitation. In family IIn, cardiological examination of a son of patient IIn showed no signs of DCM although this patient did have a left bundle branch block, which can be considered a possible precursor of DCM (see figure 2).
Cohort III: Hospital-based registry of childhood-onset patients with cancer treated with systemic cancer therapy
Six patients with AACM of the childhood cancer registry met our selection criteria (figure 1). Of these, one declined to participate and two other patients were already included in cohort I (patients Id and Ie). In family IIIa, we identified a second-degree relative of the index patient who died suddenly at the age of 56 years. In family IIIb, we identified a second-degree relative diagnosed with AF and heart failure. In family IIIc, the father of the index patient was diagnosed with arrhythmia (unspecified) and a cousin of the mother of the index patient had died unexpectedly at the age of 40 years. Detailed clinical data and relevant family data of patients IIIa, IIIb, IIIc are shown in table 1.
Genetic analysis
The genomic DNA of all index patients in DCM families with patients with AACM (cohort I, n=5) and six patients with AACM (n=3 from cohort II and n=3 from cohort III) were screened for mutations in 48 cardiomyopathy-associated genes using next-generation DNA sequencing.18 Two pathogenic mutations, six VUS1s, and three VUS2s were identified (table 2).
Sequence variants identified through next-generation DNA sequencing of 48 cardiomyopathy-associated genes
The two pathogenic mutations included a previously reported complex mutation (two missense mutations in MYH7 on one allele, c.1633G>A (p.Asp545Asn) and c.2863G>A (p.Asp955Asn)) in family Ie13 ,20 and a nonsense mutation (c.4125T>A (p.Tyr1375*) in MYH7) in family IIn. Co-segregation analysis showed that the complex mutation fully segregated with disease in family Ie.13 Co-segregation analysis of the nonsense mutation could not be performed, as there was no DNA available from the possibly affected son of the index patient (family IIn). Sanger sequencing for mutations in the PLN, MYH7, LMNA and TNNT2 genes in the remaining patients of cohort II (n=10) revealed one VUS2 in family IIc (MYH7 c.5534G>A, p.Arg1845Gln).
Discussion
This study is the first to systematically investigate the concept that a genetic/familial predisposition for DCM might be a potential risk factor for AACM. In addition to the patients identified in our initial report,13 we have identified three new DCM families with patients with AACM (families Ia, Ib and Ic) and revealed previously undiagnosed precursor signs of mild DCM in a first-degree relative of a patient with AACM (family IIn). Moreover, in two families (Ie and IIn), we identified pathogenic MYH7 mutations. These two pathogenic MYH7 mutations highlight the genetic nature of familial DCM in these families. Yet, this does not prove a causal relation between the identified MYH7 mutations and the development of AACM in these families, as AACM developed years after anthracycline treatment and other (non-genetic) risk factors might have been involved in AACM development.
In the literature, we found two other reports of patients with AACM with a positive family history of cardiomyopathy.21 ,22 Interestingly, one of these families also carried a MYH7 mutation (MYH7 c.4276G>A, p.Glu1426Lys). Hence, thus far, MYH7 mutations have been described in three of eight patients with AACM. Based on these numbers, one might speculate that MYH7 mutations are more frequent in families with DCM and AACM compared with lone DCM families.19 However, this finding is likely to be coincidental and also does not, as indicated before, prove a causal relation between the development of AACM and MYH7 mutation carriership. Given the large heterogeneity of DCM, it is plausible that genetic analysis of larger numbers of patients with AACM will probably reveal pathogenic mutations in cardiomyopathy-associated genes other than MYH7.
In addition to pathogenic MYH7 mutations, we identified several variants of unknown clinical significance (VUS1s and VUS2s). Based on current knowledge, it is not clear whether these variants are involved in the pathogenesis of DCM. It is possible that they represent only benign sequence variations. However, it is conceivable that these variants are low-risk or high-risk pathogenic alleles or modifiers. This is especially the case for the VUS2 reported in VCL (c.2969C>T, p.Ala990Val) in family Ic. This variant has not been reported before in cardiomyopathy patients or in the 1000 Genomes project. It has been reported at low frequencies in ∼6500 exomes of the exome sequencing project (ESP) of the National Heart, Lung, and Blood Institute (NHLBI) (minor allele frequency (MAF) 0,0003) and 1000 Dutch genomes of the GoNL project (MAF 0,003). In addition, the Ala990 amino acid position is highly conserved and several prediction programs classify this amino acid substitution as pathogenic. Moreover, segregation analysis showed that this variant fully segregated with the disease in this family.
Our results support the hypothesis that a genetic/familial predisposition for DCM might be a potential risk factor for AACM. The question remains: what is the potential mechanism? A conceivable mechanism could be that genetic inactivation of DCM associated proteins may lower the threshold for the damaging effect of anthracyclines on cardiomyocytes and cardiac function.23 Depending on reserve and restoration capacity this may result in AACM at a lower threshold.24
Limitations and future research
The descriptive nature of our study design did not enable us to perform statistics on our hypothesis that a genetic predisposition for DCM might be a potential AACM risk factor. Nor did it allow to statistically address the several confounders leading to AACM (such as radiotherapy or coronary artery disease). Also, given that cancer and heart disease are both common diseases of the ageing population,25 it is possible that some included patients developed heart failure independently of anthracycline treatment.26 On the other hand, cardiological evaluation and cardiac follow-up were not offered to all patients treated with anthracyclines. Since signs of AACM may only become clinically evident several years after anthracycline treatment,4 ,6 it is plausible that we have underestimated the true number of patients with AACM in our study. Further, it is conceivable that some of the patients are carriers of currently unknown pathogenic mutations because, at present, a genetic cause can be found in only ∼50% of patients with DCM.19 ,27
More research is needed to further elucidate the intriguing relationship between a potential genetic predisposition for DCM and AACM. Ideally, this should be multicentre systematic studies including next generation DNA analysis (gene panel-based and/or exome sequencing) in patients with AACM, and cardiological screening of first-degree family members. Not only will such studies add to our understanding of the aetiology of AACM and lead to the identification of patients at a high risk for AACM, they may also lead to the identification of novel targets important for the prevention of AACM or even novel cardiomyopathy susceptibility genes.
Conclusion
We have identified five DCM families with one patient with AACM, and one patient with AACM with a family member with a previously unrecognised, possible early sign of mild DCM. In two of these families, we identified pathogenic MYH7 mutations, confirming the genetic character of the DCM in these families. Although more research is needed, our current data support the hypothesis that a genetic susceptibility for DCM is a potential risk factor for AACM. In view of the reported cases, one should be vigilant for increased susceptibility to AACM if patients report a family history of DCM/heart failure and one may, in these particular cases, consider more intensive cardiovascular monitoring before and during cancer treatment or alternative non-cardiotoxic cancer therapy.
Acknowledgments
We are obliged to all patients who participated in this study. We thank Jackie Senior and Kate McIntyre for editing this manuscript, Nynke Zwart for her help with retrieving data from the childhood cancer registry, and Eddy de Boer, Lennart Johansson and Ludolf Boven for DNA analyses.
References
Footnotes
MW and KYvS-Z contributed equally.
-
Contributors The conception, design, analysis and interpretation of data and drafting of the manuscript were done by MW and KYvS-Z. NLW and JDHJ contributed to the manuscript by analysing and interpretating the data. AP, JAG, JPvT and MPvB provided critical remarks and input to the conception and design of the study. The manuscript was circulated to all the authors for comments. All the authors have seen and approved the final version of the manuscript.
-
Competing interests None.
-
Ethics approval Ethical committee of the University Medical Center Groningen .
-
Provenance and peer review Not commissioned; externally peer reviewed.
-
Data sharing statement No additional data are available.