Article Text

Abstract
The term ‘therapeutic angiogenesis’ originated almost two decades ago, following evidence that factors that promote blood vessel formation could be delivered to ischaemic tissues and restore blood flow. Following this proof-of-principle, safety and efficacy of the best-studied angiogenic factors (eg, vascular endothelial growth factor) were demonstrated in early clinical studies. Promising results led to the development of larger controlled trials that, unfortunately, have failed to satisfy the initial expectations of therapeutic angiogenesis for ischaemic heart disease. As the quest to delay the progression to heart failure secondary to ischaemic heart disease continues, alternative therapies have emerged as potential novel treatments to improve myocardial reperfusion and long-term heart function. The disappointing results of the clinical studies using angiogenic factors were followed by mixed results from the cell therapy trials. This review reflects the current angiogenic strategies for the ischaemic heart, their limitations and discusses future perspectives in the light of recent scientific and clinical evidence. It is proposed that combination therapies may be a new direction to advance therapeutic repair and regeneration of blood vessels in the ischaemic heart.
- Coronary Artery Disease
- Myocardial Ischaemia and Infarction (IHD)
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 3.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/3.0/
Statistics from Altmetric.com
Introduction
An inadequate supply of blood to the myocardium characterises ischaemic heart disease (IHD), resulting in an imbalance between myocardial oxygen supply and demand. The major causes of IHD are atherosclerosis, thrombosis or embolus in the coronary arteries that lead to low perfusion in the region supplied by the culprit vessel. Coronary artery disease (CAD) remains the primary cause of mortality worldwide.1 In addition to CAD, pathologies such as aortic stenosis and hypertrophic cardiomyopathy can also result in ischaemic damage to the myocardium.2 ,3 During the past decade, treatment for IHD has advanced into an inspiring new era. Improved regimes of medical therapy, which combine statins, antiplatelets agents, ACE inhibitors, β-blockers, and so on, offer the optimal treatment and prevention for CAD. Furthermore, percutaneous coronary intervention (PCI) and coronary artery bypass grafting (CABG) restore blood flow in the ischaemic territory and provide substantial gains for patients surviving ischaemic episodes. Revascularisation decreases the subsequent effect of acute haemodynamic instability and chronic unfavourable left ventricular (LV) remodelling, and it has contributed to a remarkable decrease in mortality.1 ,4–6 However, even treatments meeting today's highest standard of medical care are unable to halt the progression of IHD to chronic heart failure (HF), adding to its rising incidence.7 Therapeutic angiogenesis to promote myocardial perfusion and improve LV function has been the focus of extensive preclinical and clinical studies. Here, we review the main trials aimed at targeting blood vessel repair and regeneration in the ischaemic heart, and discuss the limitations of the current therapies.
Revascularisation in heart repair
Following revascularisation (eg, PCI or CABG), up to 90% of the patients suffering from occluded coronary artery would achieve almost immediate patency. Hence, the 5-year cardiac event-free survival expectancy of patients with CAD receiving primary revascularisation has increased considerably from ∼30% to >85%.8 However, it is estimated that a quarter to one-third of the patients would experience inadequate myocardial reperfusion and would face higher risks of developing ventricular remodelling and early death.8 In the last decade, no better treatment option has become available for the patients who are either not suitable for revascularisation or have not achieved a complete revascularisation. Importantly, even in patients who successfully undergo primary standard CABG, restenosis of the vein grafts is commonly seen after several years, followed by a decreased survival and recurrence of symptoms. They generally require secondary revascularisation, regardless of the normal patency of the grafted conduit. These patients, therefore, continue to experience residual myocardial ischaemia (MI) despite optimal therapy, and are in need of an alternative or supplementary revascularisation strategy to bring further attenuation or halt the advance of myocardial damage.9 ,10 As it is important to achieve appropriate myocardial collateral blood flow and microvascular perfusion, repair and/or regeneration of blood vessels has emerged as a therapeutic approach for IHD.
Blood vessel formation is characterised by (1) the sprouting of new blood vessels from the existing ones (angiogenesis) and endothelial cells are responsible for the capillary growth, migration and organisation of the vessel lumen, (2) the increase in the size of the lumen of pre-existing arterioles by remodelling and growth (arteriogenesis), a proccess that is controlled by perivascular mural cells or (3) de novo vessel formation involving the migration, differentiation and incorporation of endothelial progenitors, normally from the bone marrow, into the damaged vessels (vasculogenesis;11 figure 1). During adulthood, the quiescent vasculature is maintained in a paracrine fashion by angiopoietin-1 (Ang1) expressed by stromal and perivascular cells. Ang1 binds to its receptor Tie2 on the endothelial cells where angiopoietin 2 (Ang2) is stored (figure 1A). Neovascularisation is induced by ischaemia, cytokines and shear stress of blood flow, which causes the autocirne release of Ang2 competing with Ang1 to bind its receptor (Tie2). Such activation enables the endothelial cells to respond to exogenous growth factors (figure 1B). During angiogenesis, cytokine receptors on the surface of endothelial cells are activated by exogenous vascular endothelial growth factor (VEGF), fibroblast growth factor (FGF), insulin growth factor (IGF) and transforming growth factor (TGF), inducing signalling pathways that modulate cell survival, proliferation and migration as well as vascular permeability. Activated endothelial cells produce platelet-derived growth factor (PDGF), which induces proliferation of the stromal cells via the Erk1/2 signalling pathway and further release of angiogenic factors (figure 1C). In addition to producing angiogenic growth factors, perivascular stromal cells are responsible for the extracellular matrix (ECM) remodelling. These cells produce metalloporteinases (MMPs), plasminogen activator (PA) and collagenases to cleave ECM components and to facilitate the migration of endothelial cells. This process is balanced by the secretion of protease inhibitors such as tissue inhibitor of metalloproteinases (TIMP) and PA inhibitor by stromal cells. Vessel maturation is controlled by the perivascular stromal cells (figure 1C), allowing the enlargement of vessel lumen. The increment in blood flow restores the paracrine effect of Ang1-Tie2 signalling followed by the maintenance of vascular quiescence (figure 1D). Recruitment of bone marrow progenitor cells to the ischaemic tissue, or vasculogenesis, is also observed, and it is another mechanism of neovascularisation in response to ischaemia (figure 1E). Therapeutic angiogenesis aims at restoring the microcirculation in the ischaemic myocardium by delivering proangiogenic factors. The first promising results in humans were reported in severely symptomatic patients with critical limb ischaemia where VEGF was administered directly into the skeletal muscle, increasing the formation of collateral vessels.12 This followed experimental proof of the concept in rabbits with induced unilateral limb ischaemia which received intra-arterial infusion of an acidic fibroblastic growth factor (aFGF or FGF1) encoding vector.13 Soon after, clinical trials were also extended to patients with advanced symptomatic CAD14 and those not eligible for standard revascularisation strategies.15 In the first instance, patients with three vessel disease received human recombinant FGF1 (hrFGF1). Formation of capillaries could be observed in all patients around the site of injection.14 In the second trial, plasmid DNA expressing VEGF165 directly injected into the myocardium was demonstrated to be safe and led to reduced symptoms and improved myocardial perfusion in some patients.15

Process and mechanisms of blood vessel formation. (A) Vascular quiescence is maintained by the Ang1-Tie2 signalling pathway. Ang1 is expressed in perivascular SC and binds to the Tie2 receptor on EC in a paracrine fashion to stabilise the vasculature. EC in turn store Ang2. (B) Vascular activation is induced by multiple factors, such as hypoxia, VEGF and shear stress of blood flow on the vascular wall, and Ang2 is released from EC and competes with Ang1 to bind the Tie2 receptor. The autocrine antagonising effect of Ang2 on Ang1-Tie2 signalling activates the ECs by enabling them to react to growth factors, such as VEGF and FGF. EC that lack Ang2 production are likely to fail to respond to exogenous growth factors. (C) Angiogenesis is a crucial mechanism and process for neovascularisation where vascular sprouting and elongation take place. The angiogenesis process relies on the EC-SC interaction and it is fine-tuned through growth factor signalling pathways and remodelling of the ECM. Activated EC express cytokine receptors which respond to exogenous VEGF, FGF, IGF and TGF-α signalling to promote cell survival, cell migration, cell proliferation and vascular permeability. Simultaneously, activated EC produce PDGF which binds to PDGFR-β on SC to promote their proliferation via the Erk1/2 signalling pathway. SC play a critical role in ECM remodelling during angiogenesis besides the secretion of angiogenic growth factors. Proteinases such as MMP, PA and collagenase are released from the SC to cleave ECM proteins to facilitate cell migration and vascular elongation. The process can be interrupted by TIMPs and PAIs, which target and antagonise MMP and PA. (D) Vascular maturation is seen as the neovasculature start sustaining a regular blood flow. The tissue reperfusion enables the overexpression of Ang1, and the restored Ang1-Tie2 signalling in turn suppresses Ang2 production and encourages Ang2 storage, stabilising the vascular cells while increasing the vascular diameter. (E) Vasculogenesis is another mechanism of neovascularisation. The mobilised progenitor cells from bone marrow penetrate the ischaemic tissue and incorporate with the newly formed local vascular network. Ang1, angiopoietin1; Ang2, angiopoietin 2; EC, endothelial cells; ECM, extracellular matrix; FGF, fibroblast growth factor; IGF, insulin growth factor; MMP, metalloproteinase; PA, plasminogen activator; PAI, plasminogen activator inhibitor; PDGF, platelet-derived growth factor; PDGFR-β, platelet-derived growth factor receptor β;SC, stromal cells; TGF-α, transforming growth factor-α TIMP, tissue inhibitor of metalloproteinase; VEGF, vascular endothelial growth factor.
By contrast, antiangiogenic cancer therapies have resulted in cardiac toxicity. Drugs such as bevacizumab (a specific VEGF-blocking antibody), sorafenib and sunitinib (both tyrosine kinase inhibitors) have been used against tumour angiogenesis.16 Although with different molecular targets and different specificity, the use of these approved drugs has been associated with cardiovascular side effects such as decrease in left ventricular ejection fraction (LVEF), increased incidence of HF, hypertension and MI.17 ,18
Angiogenic factors for IHD
A number of proangiogenic factors are expressed in the ischaemic myocardium.19 ,20 However, the VEGF and FGF families are the best-studied growth factors in angiogenesis following MI and the only ones that have been tested in the clinical setting (see table 1).
Randomised controlled trials with proangiogenic factors
The VEGF family
The VEGF family of angiogenic cytokines includes VEGF-A, VEGF-B, VEGF-C, VEGF-D and placental growth factor (PlGF) in humans. They have high affinity for their receptors, namely VEGFR1 and VEGFR2, on endothelial cells which results in the activation of signalling cascades and the production of nitric oxide (NO) by these cells.21 ,22 Two different isoforms of VEGF-A have been tested in clinical trials of VEGF-A165 (containing 165 amino acids) and VEGF-A121 (a shorter form of VEGF-A consisting of 121 amino acids). The role of VEGF-B and PlGF in angiogenesis still remains controversial and their clinical efficacy has not been assessed yet.23 VEGF-A production is significantly upregulated by ischaemia in pigs and rats,24 ,25 suggesting that VEGF-A is a likely mediator of ischaemia-induced myocardial neovascularisation. Preclinical studies showed that administration of an adenoviral vector expressing VEGF (AdVEGF) improved capillary density and LV function in a rodent model of acute MI (AMI).26 In a large animal model of chronic MI, the injection of AdVEGF121 into segments of stunned myocardium increased collateral vessel flow and improved cardiac function.27 In addition to these experimental studies, early clinical trials with promising results led to the development of randomised controlled trials (RCTs) where recombinant protein and plasmid DNA or adenoviral vectors expressing the angiogenic factors were used for treatment (table 1).
In the phase II VIVA trial, patients with stable CAD and no revascularisation option received an intracoronary infusion followed by intravenous injection of placebo or two different doses of recombinant human VEGF (rhVEGF-A165) protein.28 There were no significant differences in exercise tolerance, quality of life (QoL) or myocardial perfusion at 60 days between the treated and placebo groups. At 120 days, a high dose of rhVEGF-A165 improved the angina class only and, hence, the trial was terminated prematurely. In the EUROINJECT-ONE29 and NORTHERN30 trials, patients with severe stable IHD and no other treatment option received a percutaneous intramyocardial injection of plasmid DNA expressing VEGF165 (phVEGF-A165). Neither of these trials found significant improvement in myocardial perfusion in the treated groups compared with the placebo groups despite some improvement in wall motion and LV function in the EUROINJECT-ONE trial.29 ,30 Moreover, the REVASC study enrolled patients with intractable angina and no option of revascularisation to receive either AdVEGF-A121 or maximal medical therapy. Interestingly, there were significant improvements in exercise tolerance and QoL in the AdVEGF-A121-treated patients compared with controls.31 However, the placebo effect cannot be ruled out in this trial as patients in the control arm did not have a thoracotomy or receive placebo. The NOVA double-blinded, placebo-controlled, multicentre study investigated the safety of intramyocardial injection and the efficacy of BIOBYPASS (AdGVVEGF121.10NH) gene therapy in patients with refractory advanced CAD.32 Injection of AdGVVEGF121 did not improve exercise capacity or myocardial perfusion in a 52-week follow-up, and this study was also terminated prematurely. Finally, the 8-year follow-up of the Kuopio angiogenesis trial (KAT) showed that VEGF-A165 expressed transiently in either an adenoviral vector or a plasmid was safe, did not increase the risk of mortality and it was well tolerated.33 ,34 Nonetheless, no significant improvements were observed in the treated patients compared with the control patients. In the AdVEGF-A165 group, myocardial perfusion improved compared with the plasmid VEGF-A165 group at 6 months.34
The FGF family
The FGF family comprises up to 22 ligands and four tyrosine kinase receptors, adding to the complexity of this system. Several of its members have been reported to promote angiogenesis in preclinical studies (for review, please see35). The best studied members of the family are FGF-1 and FGF-2, and together with them, FGF-4 has been tested in clinical trials to improve angiogenesis post-MI. The safety and feasibility of recombinant FGF-2 (rhFGF-2) in therapeutic angiogenesis have been evaluated in patients with chronic MI during CABG36 and in the FIRST trial in participants with stable angina pectoris secondary to CAD.37 While the phase I trial by Laham et al36 delivered a single intracoronary injection of rhFGF-2 and showed signs of therapeutic efficacy, the larger phase II FIRST trial37 that recruited 337 patients did not reveal any significant improvement in exercise tolerance or ischaemic areas in treated patients. Only a significant improvement in angina class and angina frequency was observed in the more affected patients.37 The phase I KAT301 study is an ongoing trial designed to evaluate the safety and efficacy of intramyocardial injection of AdVEGF-D in patients with chronic ischaemia and no option of revasvularisation (http://clinicaltrials.gov/show/NCT01002430).
Like VEGF, FGF gene transfer was based on the use of adenoviral vectors as gene transfer vehicle. In the Angiogenic GENe Therapy (AGENT) double-blind RCT, the administration of five increasing doses of Ad5-FGF4 in patients with stable angina pectoris showed a trend towards clinical improvement in exercise tolerance. A single intracoronary administration of Ad5-FGF4 was safe and well tolerated with no immediate adverse events.38 The efficacy of Ad5-FGF4 therapy was further examined in the AGENT 2, AGENT 3 and AGENT 4 studies. The AGENT 2 study showed an encouraging trend for improved myocardial perfusion in treated patients compared with the placebo group,39 whereas the results of the AGENT 3 and AGENT 4 trials showed no significant differences between treatment and placebo in angina symptoms, angina class or exercise tolerance. However, when stratified by gender, the data indicated that in women, the placebo effect was small and the treatment had a significant effect.40
The reparative and regenerative function of stem cell therapies
Stem cell therapies have not been purely directed at stimulating blood vessel growth in the ischaemic heart but rather to repair and regenerate all cardiac tissues in their capacity of multipotent cells. The first evidence that stem/progenitor cells have the potential to be used as treatment for IHD was reported by Orlic et al in 2001. In this study, haematopoietic stem cells (HSC) mobilised into circulation and injected into infarcted myocardium of mice were able to improve heart function and regenerate heart tissue.41 Since then a number of cell therapies have been tested in clinical trials. Here, we review trials that have administered cell therapies with the aim to improve a long-term heart function and myocardial perfusion.
Bone marrow mononuclear cells
Unselected bone marrow mononuclear cells (BMNC) are clearly the most investigated cell-based therapy for IHD in clinical studies, with the longest follow-up lasting up to 5 years.42 ,43 An attractive novel treatment for acute and chronic MI, BMNC are relatively easy to harvest, easy to process in a short time-frame using standardised techniques (eg, density gradient centrifugation and cell sorting) that usually yield large quantities of cells, ready to be administered to the patients in a matter of hours if required. This makes them extremely amenable to treat patients with AMI. In this patient cohort, BMNC have a beneficial but moderate effect on heart function.42 ,43 Not surprisingly, following the expectations raised by the early-phase small clinical studies, several RCTs have generated mixed results (table 2).
Major cell-based therapy randomised controlled trials
The largest trial, the REPAIR-AMI, recruited patients post-AMI and showed an improvement of global LVEF in the treatment group compared with the control group (ΔLVEF=2.9%), without significant changes of LV end-diastolic volumes 4 months following cell transplantation.44 In addition, decreased mortality was observed in the treatment group compared with the control group at 2 years of follow-up.45 In contrast, in other landmark studies, BMNC have not shown the alleged beneficial effect in the same patient cohort. The ASTAMI trial did not show a significant improvement in LV function or dimensions at 4–6 months of follow-up,46 while the Belgium trial reported mixed results where there was no improvement on LVEF despite the significant reduction in infarct size,47 and the BOOST trial showed a transient effect of BMNC on LVEF.48 ,49 Moreover, RCTs such as the HEBE,50 BONAMI,51 FINCELL52 and TIME53 ,54 show no significant effect on heart function or contractility between treated and non-treated patients (table 2). Recent systematic reviews and meta-analysis, which also included smaller trials, have suggested that BMNC improve LVEF by 3–5%.42 ,43 ,55 However, there is no significant reduction on the risk of mortality in patients treated with BMNC compared with controls.42 ,43 The BAMI trial is so far the largest ongoing international multicentre RCT. It is designed to recruit 3000 patients to define the effect of single dose of intracoronary administration of BMNC on patients with AMI after successful primary revascularisation. The primary outcomes to be measured are long-term all-cause mortality, cardiac death, major associated cardiac events (MACE) and rehospitalisation between the cell therapy group and the placebo group (http://clinicaltrials.gov/show/NCT01569178).
Fewer data from RCTs are available in patients with chronic MI and HF (table 2). Intracoronary delivery of BMNC during CABG resulted in significant changes in LVEF and exercise tolerance in favour of the treatment.56 Patients with HF, receiving optimal medical treatment and with no option of revascularisation, have been treated in two other trials. Following the promising results of the phase I trial, Perin et al treated patients with HF with BMNC in a phase II trial. Surprisingly, no significant improvement in left ventricular end-systolic volume or maximal oxygen consumption was observed in treated patients compared with controls.57 The phase II FOCUS-CCTRN delivered BMNC by a percutaneous intramyocardial injection in patients with congestive HF with no option of revascularisation.57 At 6 months of follow-up, no difference in LV systolic function, myocardial perfusion or myocardial viability was observed between treated and control patients. However, a modest improvement in LVEF was apparent in patients aged <62 years, although not in the treated group in its totality. In another trial, intramyocardial delivery of BMNC to patients with ischaemic HF improved HF symptoms, and LV function and even improved survival of these patients significantly.58 In Pokushalov's study, patients suffer from a more severe stage of ischaemic HF (NYHA class III–IV) with lower rates of survival compared with Perin's study (NYHA class II–IV), which may be a determinant of the decreased mortality observed in this patient cohort.
Recently, results from the CELLWAVE trial have been reported.59 The phase I/II trial evaluated the effect of intracoronary infusion of BMNC in combination with extracorporeal shock wave in patients with HF due to ischaemic cardiomyopathy. The administration of BMNC aided by shock wave showed a modest but significant improvement in LVEF and regional wall thickening 4 months following treatment. In addition, the combination treatment seems to protect against major adverse cardiac events (MACE).59
Notably, BMNC are a heterogeneous cell population that contains haematopoietic stem and progenitor cells (HSC/HPC) and endothelial progenitor cells (EPC; around 2–4%), mesenchymal progenitor cells (MSC; 0.001–0.01% of the nucleated cells), committed progenitor cells and their differentiated progeny.60 As a consequence, it is difficult to ascertain which cell types in the whole population are more potent or responsible for the therapeutic effect. This has led to the development of clinical trials which use selected cell populations such as HSC or MSC as treatment for IHD.
Haematopoietic stem cells
Human HSC, identified by CD133, CD34 or aldehyde dehydrogenase (ALDH) markers, have been tested in clinical trials as treatment for IHD. CD133+ or CD34+ HSC can be enriched from mononuclear cells, either from the bone marrow or mobilised peripheral blood, by immunomagnetic cell separation procedures.61 ,62 ALDH is a cytosolic enzyme used to identify stem cell populations. High ALDH activity has been found in the CD34+ lineage- (Lin-) HSC compartment and identifies primitive HSC. ALDH+ cells are isolated from the bone marrow by cell sorting.63 Enriched CD133+ HSC were first injected into the infarct border zone of patients undergoing CABG.61 Promising results from this small trial, with no adverse events and improved LV function and myocardial perfusion, led to the development of larger RCTs (table 2). Recently, the REGENT trial compared CD34+ CXCR4+ HSC with unfractionated BMNC or control in patients who suffered from AMI and received PCI as primary intervention. However, no significant improvement in LV function was observed between trial arms.62 Patients undergoing off-pump CABG and presenting LV dysfunction (LVEF <35%) were treated with bone marrow CD34+ HSC in a small RCT.64 The study showed a significant improvement in LVEF in treated patients compared with controls. In addition, CD34+ HSC have been tested in a phase I trial in patient with refractory angina and no option of revascularisation. In this study, the patients received GCS-F to mobilise bone marrow cells into circulation and CD34+ cells were enriched from peripheral blood mononuclear cells prior to injecting them into the myocardium using electromechanical mapping. Unlike in AMI trials, the primary outcome measured by this trial was change in angina frequency.65 Administration of CD34+ cells resulted in a significant reduction of angina frequency (the number of angina episodes per week). The results were confirmed in a dose escalation phase II trial66 and this has been followed by the first phase III clinical trial with CD34+ cells as treatment for patients with refractory angina which is currently ongoing.67 Recently, a newly identified HSC population expressing the ALDH enzyme has been tested in patients with chronic ischaemia and no option of revascularisation. Perin et al63 provided preliminary evidence that treatment with ALDH+ cells is safe and may improve perfusion in those patients.
Mesenchymal stem/stromal cells
MSC constitute another potential option for stem/progenitor cell-based therapy. MSC express cell surface markers such as CD90, CD105, CD44 and CD73, but not the haematopoietic markers CD45, CD34, CD14, CD19 and HLA-DR, and due to their immunomodulatory properties68 they have attracted much commercial interest as they can be generated as an ‘off-the-shelf’ product for allogeneic cell transplantation. They have been identified in tissues such as bone marrow and adipose tissue. Autologous and allogeneic MSC transplantation is currently under investigation for IHD among other conditions; however, clinical data are scarce (table 2).
The first RCT using autologous BM-derived MSC treated patients who had suffered AMI and underwent PCI. The MSC treatment proved to be safe and significantly improved LV function.69 Autologous BM MSC also improved exercise tolerance, NYHA class and LVEF significantly in patients with severe ischaemic cardiomyopathy.70 A safety double-blinded phase I trial with intravenous administration of allogeneic BM MSC to treat patients with AMI showed a significant improvement in LVEF compared with controls.71 Following this the authors compared autologous and allogeneic MSC in patients with ischaemic cardiomyopathy.72 In the phase I/II POSEIDON trial, three doses of MSC were injected into patients. However, no control group was included in this trial. Autologous, but not allogeneic MSC, improved exercise capacity and QoL significantly. None of the injected cells improved maximal oxygen consumption or LVEF. In contrast, allogeneic MSC significantly reduced LV end-systolic volume. In this early study, no immunological reaction to MSC therapy was observed.72 It would be interesting to see the results of the ongoing MSC-HF trial, a double blind, placebo-controlled trial administering BMSC to patients with chronic ischaemic HF.73 Recently, BMSC preconditioned by exposure to a cardiogenic cocktail have been delivered by percutaneous intramyocardial injection to patients with HF and no option of revascularisation in the C-CURE phase I/II trial.74 Compared with control patients, treated patients showed a significant improvement in LV systolic function and symptom relief, as well as a reduction in the rate of rehospitalisation and event-free survival.
Recently, adipose tissue MSC have been tested in the clinic in patients with IHD. The APOLLO and PRECISE trials showed that freshly isolated adipose tissue-derived MSC is a safe treatment in patients suffering from AMI and chronic MI (table 2). MSC infusion resulted in a significant improvement of perfusion defect and a 50% reduction of scar size in patients with AMI but there was no improvement in LV function after 6 months.75 Similarly, in patients with no option of revascularisation, cell transplantation showed a moderate decreased in scar size at 6 months and a significant increase of peak oxygen consumption at 18 months, but no improvement of LV function was reported.76
Cardiac stem/progenitor cells
The human heart has been considered a terminally differentiated organ until recently, with no capability for self-repair. However, an elegant study by Bergmann et al77 showed the first evidence of human cardiomyocyte renewal during adult life time, suggesting that the heart contains a population of stem/progenitor cells which may be involved in the steady-state tissue repair that occurs under normal and pathological conditions. These cardiac stem/progenitor cells (CSC) are characterised by the expression of c-kit,78 Sca-179 or Islet-180 cell surface markers, the cardiac side population cells81 or the formation of clusters called ‘cardiospheres’.82 ,83
The first clinical trials using CSC as treatment for MI, the CADUCEUS and SCIPIO trials, have now been completed and have demonstrated that autologous transplantation of CSCs is safe.84 ,85 In the CADUCEUS trial, patients who have suffered from a recent MI received an intracoronary injection of cardiosphere-derived cells or placebo. The results showed a significant improvement in myocardial viability and a reduced scar size 4–6 months following cell transplantation. Interestingly, the reduction in infarct size was not accompanied by an improvement in heart function and contractility.85 In the SCIPIO trial, end-stage HF patients with LVEF <40% who did not respond to bypass surgery received an intracoronary infusion of c-kit+ CSC. The results confirmed a significant reduction in infarct size and increase in LVEF in the treated group compared with the control group.85
Limitations of the current therapies and future perspectives
The promising results obtained in preclinical models have not been replicated successfully in the clinic. The angiogenic response to ischaemia might be impaired in patients at multiple levels and influenced by a combination of cardiovascular risk factors and the hostile microenvironment in the heart, which are absent in the experimental models. In the ischaemic human heart, there might be a decreased production of factors that would be able to promote vessel sprouting and vessel maturation. There might also be a decreased number of cells with pro-angiogenic properties expressing receptors for those factors. Additionally, cells may have an impaired ability to home to ischaemic tissues and engraft, to promote vascular repair. Finally, if we believe that inflammation is, to some extent, beneficial for tissue repair and blood vessel formation, the current medical therapies, which include anti-inflammatory drugs, may have a detrimental effect on the process of angiogenesis and collateral vessel growth.
Angiogenic therapies
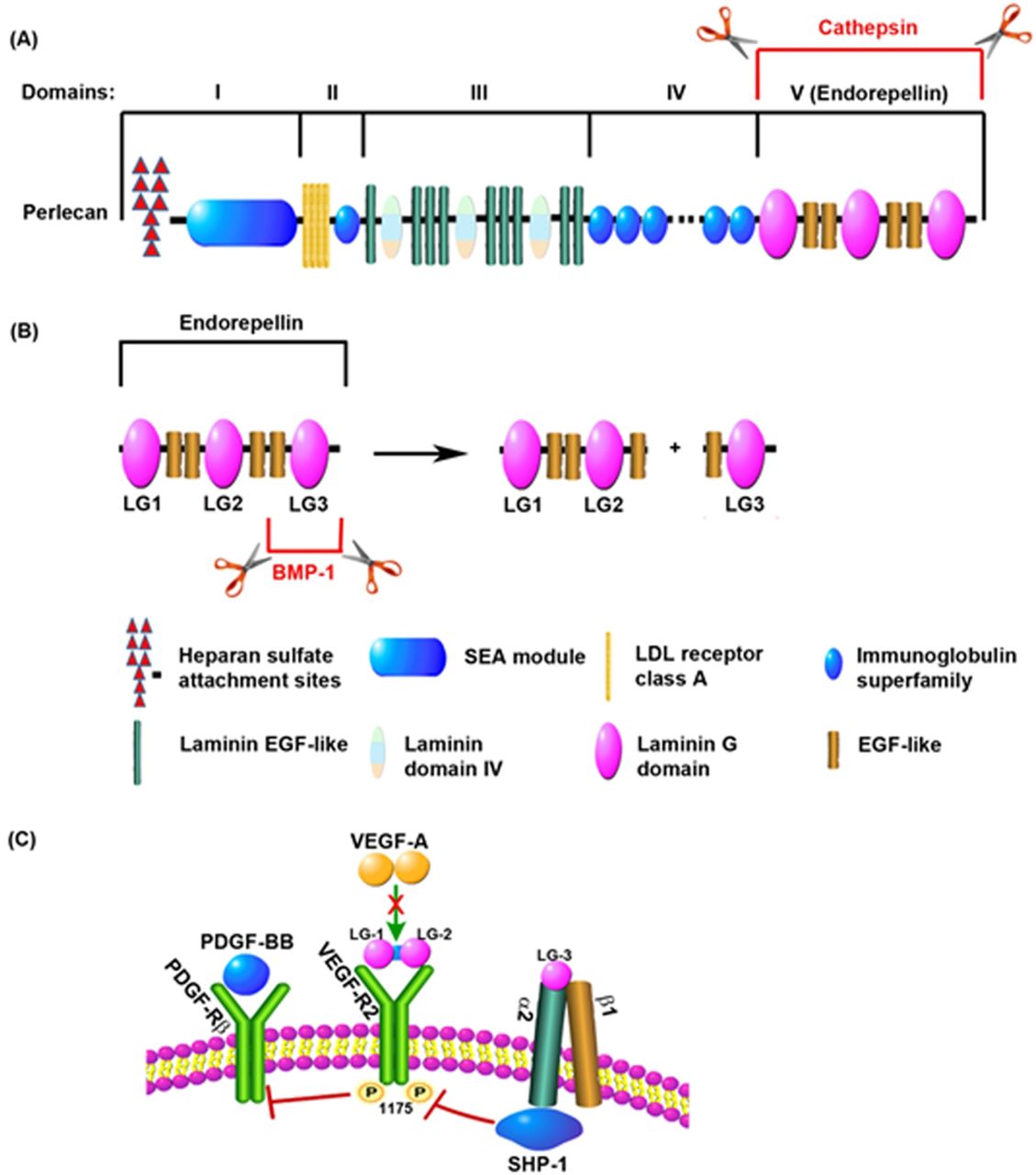
The disappointing results of the pro-angiogenic factor trials might be explained by inadequacies in the current approaches as the main obstacle remains; an inability to deliver an effective angiogenic stimulus to the ischaemic human heart. Either recombinant protein with a relative short half-life or a transient expression in a plasmid or a viral vector that does not integrate into the host cell genomes and exert low transfection and transduction efficiencies in the target tissue and proangiogenic factor monotherapies have all failed to fulfil the early promises of therapeutic angiogenesis. Furthermore, an inadequate route of delivery and suboptimal doses may have diminished the efficacy of the treatment. Mechanistically, VEGF and FGF might not be the ideal therapeutic candidates to be used, or at least not on their own but in combination with arteriogenic factors. In addition to its pro-angiogenic action, VEGF has a major role in the maintenance of vascular integrity via an endothelial-specific autocrine mechanism that promotes cell survival86 (figure 2). In mice, endothelial-specific deletion of VEGF results in vascular degeneration that cannot be rescued by exogenous VEGF.86 This may have major implications for patients with CAD and specially those suffering from diabetes, as it is a common cardiovascular risk factor characterised by endothelial cell dysfunction.87 Angiogenic factor monotherapies might not be able to salvage the ‘degenerating’ endothelium in patients with CAD. Moreover, VEGF can also be artherogenic and hence detrimental as increased microvessel density in the arterosclerotic plaque is associated with disease progression.88 ,89 Recently, a great deal of heterogeneity between endothelial cells from different sources has been revealed90 and this may have to be taken into consideration when designing future trials targeting angiogenesis in IHD. It is also plaussible that the effect of exogenous VEGF on its receptors is inhibited by endogenous antagonists released from the ECM by MMPs (figure 3). One of these antagonists is endorepellin, the C-terminal or V domain of perlecan, released by Cathepsin-L (figure 3A), which has an inhibitory effect on VEGF signalling.91 Endorepellin contains three laminin G domains, named LG1, LG2 and LG3 (figure 3B). It is known that the metalloproteinase bone morphogenetic protein-1 cleaves the LG3 fragment of endorepellin92 causing an increased activity of the molecule by making the two proteolytic products (LG1-LG2 and LG3) available to act separately. LG1-LG2 binds VEGF-R2 directly competing with VEGF for its receptor, while LG3 binds to α2β1 integrin, which also inhibits the VEGF-mediated activation of VEGF-R293 ,94 (figure 3C). Indirectly, high levels of these proteinases could have an antiangiogenic effect.
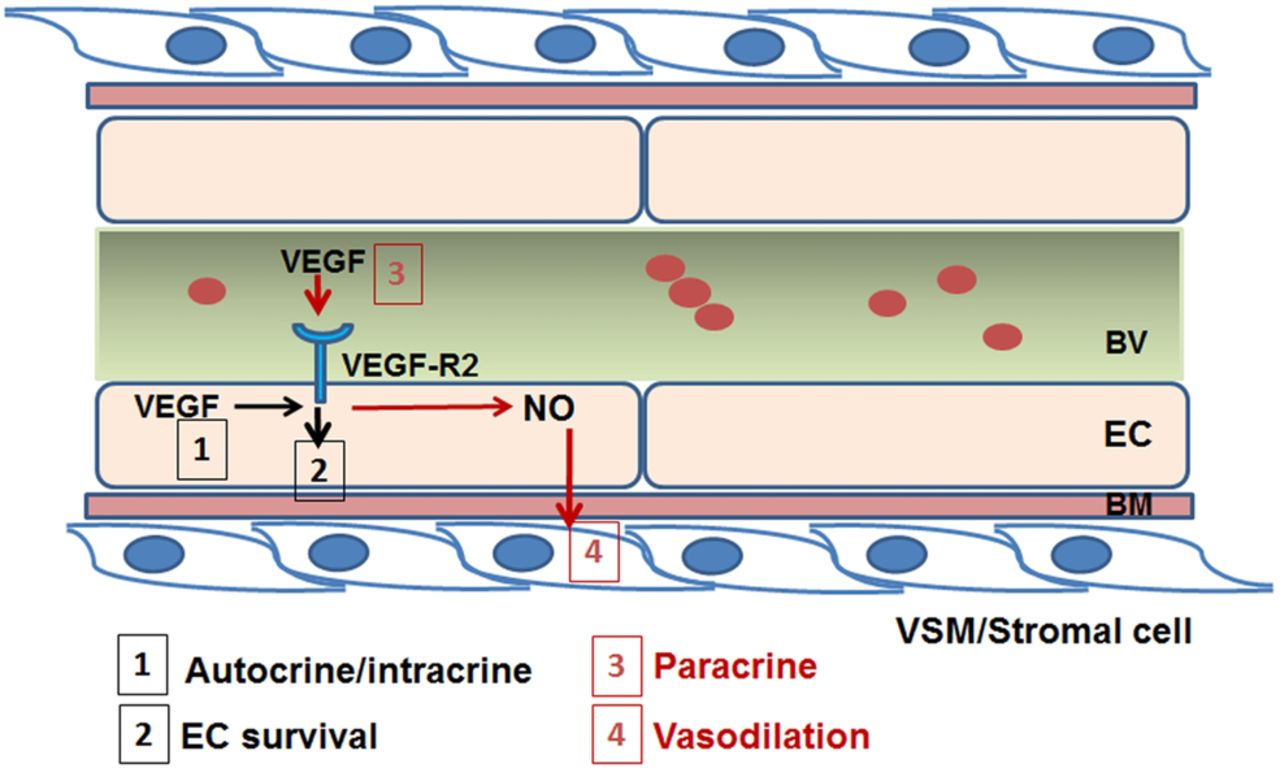
The role of vascular endothelial growth factor in vascular homoeostasis. In the adult vasculature, VEGF produced by endothelial cells acts by an autocrine/intracrine mechanism (1) activating its receptor, VEGF-R2 and activating signalling pathways involved in endothelial cell survival (2). Moreover, VEGF produced by mural cells in the perivascular niche or circulating in blood vessels have a paracrine action (3) inducing the production of nitric oxide and causing vasodilation and inhibition of proliferation of vascular smooth muscle cells (4). BM, basal membrane; BV, blood vessel; EC, endothelial cells; NO, nitric oxide; VEGF, vascular endothelial growth factor; VEGF-R2, vascular endothelial growth factor receptor 2.

{kind=link}
{kind=link}
{kind=link}
Inhibition of VEGF-R2-mediated signalling cascade by endogenous antagonists. (A) Perlecan, an extracellular matrix component, is composed of several functional domains. The C-terminal domain, also named V domain or endorepellin, is released from the full length protein by proteolytic processing by cathepsin-L. (B) Endorepellin contains three laminin G domains (LG1, LG2 and LG3) separated by EGF domains. Metalloproteinases such as BMP-1 cleave endorepellin to release the LG3 domain from the LG1-LG2 fragment. (C) The LG1-LG2 fragment of endorepellin binds to VEGF-R2 inhibiting its downstream phosphorylation. LG3 binds to the α2 domain of the α2β1 integrin which can also inhibit VEGF-R2 activation through SHP-1. Inhibition of VEGF-R2 signalling enables PDGF-Rβ-mediated signalling. BMP-1, bone morphogenetic protein-1; EGF, endothelial growth factor; PDGF-BB, platelet derived growth factor b; PDGF-Rβ, platelet-derived growth factor receptor β; SHIP-1, SH2 domain-containing inositol-5’-phosphatase 1; VEGF-A, vascular endothelial growth factor a; VEGF-R2, vascular endothelial growth factor receptor 2. Adapted from Zoeller et al.94
Arteriogenic therapies
As arteriogenesis is also required to maintain functional collateral growth, mural cells such as vascular smooth muscle cells and pericytes may need to be targetted to promote vessel maturation and stabilise collateral networks. Angiogenic cytokines are not the only factors produced during ischaemic episodes. Following MI, there is an increase in the release of MMPs produced by neutrophil infiltration in the ischaemic heart. These MMPs play a major role in the tissue remodelling that takes place post-MI as they cleave components of the ECM (figure 1). The degradation of the ECM is followed by the deposition of a new basement membrane by the mural cells. During this shift, MMP inhibitors, such as TIMPs, compensate the action of MMPs by causing the deposition of de novo-synthesised ECM components, and the junctions between the endothelial cells are then re-established. We hypothesise that an imbalance in MMP and/or TIMP production may disrupt the process of angiogenesis in the ischaemic heart. It has been reported previously that plasma levels of MMP-9 correlate with LV dysfunction following MI, while MMP-2 levels are inversely correlated to LV volumes.95 Additionally, the active form of MMP-9 and the MMP-9/TIMP ratio is significantly higher in patients with stable CAD than in healthy control individuals.96
Recently, mobilising agents such as granulocyte-marcropahge colony stimulating factor and granulocyte colony stimulating factor (G-CSF) have been used in three controlled trials to promote arteriogenesis in patients with CAD.97–99 It is proposed that G-CSF stimulate arteriogenesis by the release of monocytes and EPC.100 These promising results may have a great impact in the design of future trials using combination therapies. G-CSF has been administered previously together with cell therapies in patients with refractory angina and no other treatment option.65 ,66 However, in that setting, it is difficult to distinguish the effect of the cytokine from the effect of the cell treatment, and the trials were not designed exclusively to improve blood vessel formation.
Cell therapies
The divergent results of the cell-based therapy trials are not surprising either. Although the definite reasons for these mixed results remain largely elusive, differences in cell-related and patient-related factors are most likely to be the cause. Differences in study protocol and design, including time from reperfusion to cell injection, route of delivery, cell type, cell dose and cell isolation techniques are all factors that could influence the treatment outcome. In addition, the methods used for assessing outcomes (eg, echocardiography, MRI, LV angiography, etc) and the relevant primary outcomes should be carefully selected as each of them have their own limitations. Indeed, the majority of the trials measured LVEF as a gold standard surrogate outcome for heart function. However, a closer correlate for collateral growth would be myocardial perfusion. Ten cell therapy trials measured myocardial perfusion, but only five of them reported a significant improvement,63 ,69 ,70 ,75 ,76 and surprisingly four of these trials delivered MSC. Therefore, it is tempting to suggest that MSC might be potentially more proangiogenic and proarteriogenic than BMNC or CD34-enriched and CD133-enriched HSC. The mechanism of how cell therapies work is still not fully understood but it is suggested to be paracrine, and therefore supportive. MSC produce key factors such as Ang2, HGF, IGF-1, bFGF, SDF-1 and VEGF involved in both angiogenesis and arteriogenesis. The secretion of Ang2 by MSC and the binding to its receptor Tie2 on the EC would activate a number of receptors on the EC surface ready to respond to other angiogenic cytokines produced by MSC, thus promoting neovascularisation (figure 1). In addition, trials using cardiac progenitor cells have reported a large treatment effect on scar size,84 an outcome that has not been widely used as a surrogate of heart function or blood vessel formation. In a head-to-head comparison with bone marrow mesenchymal/stromal cells (BMSC), adipose tissue MSC and BMNC, cardiac progenitors have shown a greater therapeutic ability in a rodent model of MI, as they produce large amounts of angiogenic cytokines and possess greater myogenic and angiogenic differentiation potentials.101 Compared with the angiogenic monotherapies, cell therapies may be a more effective treatment for IHD because they provide a ‘cocktail’ of cytokines that would complement those endogenously produced to promote blood vessel formation and maturation, cell survival and/or activation of endogenous tissue repair and regeneration. However, the current cell therapies will still be facing the same challenges as angiogenic monotherapies if (1) the number of cells and cell function are affected by disease and cardiovascular risk factors leading to reduced homing capacity, (2) exogenous cytokines produced by the transplanted cells cannot rescue endothelial dysfunction (see figure 2) or (3) endogenous antagonists of angiogenic signalling pathways (figure 3) are still produced and not dampened down by cell transplantation.
Are we closer to achieving efficacy of therapeutic blood vessel regeneration and repair in IHD? Trials with angiogenic monotherapies have largely shown no effect, whereas arteriogenic factors have shown promising early results and cell therapies have shown a moderate effect on LVEF and scar size, myocardial viability and perfusion in acute and patients with chronic MI.42 ,43 ,102 Furthermore, in patients receiving maximal medical care who were not eligible for revascularisation, cell therapies significantly reduce the risk of mortality.102 Some trial results suggested that patients with a more severely impaired LV function may have a benefit from cell transplantation, whereas patients with a rather preserved LV function post-MI may not.44 Importantly, cell function is known to be affected by cardiovascular risk factors.103 ,104
Taken together, the current clinical evidence suggest that cell therapies may be a more effective treatment for IHD as they already provide a combination of cytokines that would promote blood vessel formation and maturation (angiogenesis and arteriogenesis), cell survival and/or activation of endogenous tissue repair and regeneration. However, in cases where cell function is impaired, a combination therapy (eg, cells and biologicals) that could promote arteriogenesis and/or vessel maturation rather than capillary formation only may be more suitable. Finally, patient stratification may be a pre-requisite for successful therapeutic angiogenesis.
Acknowledgments
The authors would like to thank Dr Sarah Hale for critically reading the manuscript and Professor DP Taggart and Professor SM Watt for their continuous support.
References
Footnotes
-
Contributors HZ and EM-R was involved in conception and design; HZ, CvO, DS and EM-R were involved in acquisition and interpretation of the data, and drafting of the article and approval of the final version.
-
Funding This work was supported by a National Institute of Health Research (NIHR) UK programme grant to NHSBT (RP-PG-0310–1001) and NHSBT Trust Fund (TF025).
-
Competing interests None.
-
Provenance and peer review Not commissioned; externally peer reviewed.
-
Data sharing statement No additional data are available.