
Abstract
Aims/hypothesis
Glucagon-like peptide-1 (GLP-1), a member of the proglucagon-derived peptide family, was seen to exert favourable actions on cardiovascular function in preclinical and clinical studies. The mechanisms through which GLP-1 modulates cardiovascular function are complex and incompletely understood. We thus investigated whether the GLP-1 analogue, liraglutide, which is an acylated GLP-1, has protective effects on vascular endothelial cells.
Methods
Nitrite and nitrate were measured in medium with an automated nitric oxide detector. Endothelial nitric oxide synthase (eNOS) activation was assessed by evaluating the phosphorylation status of the enzyme and evaluating eNOS activity by citrulline synthesis. Nuclear factor κB (NF-κB) activation was assessed by reporter gene assay.
Results
Liraglutide dose-dependently increased nitric oxide production in HUVECs. It also caused eNOS phosphorylation, potentiated eNOS activity and restored the cytokine-induced downregulation of eNOS (also known as NOS3) mRNA levels, which is dependent on NF-κB activation. We therefore examined the effect of liraglutide on TNFα-induced NF-κB activation and NF-κB-dependent expression of proinflammatory genes. Liraglutide dose-dependently inhibited NF-κB activation and TNFα-induced IκB degradation. It also reduced TNFα-induced MCP-1 (also known as CCL2), VCAM1, ICAM1 and E-selectin mRNA expression. Liraglutide-induced enhancement of nitric oxide production and suppression of NF-κB activation were attenuated by the AMP-activated protein kinase (AMPK) inhibitor compound C or AMPK (also known as PRKAA1) small interfering RNA. Indeed, liraglutide induced phosphorylation of AMPK, which occurs through a signalling pathway independent of cyclic AMP.
Conclusions/interpretation
Liraglutide exerts an anti-inflammatory effect on vascular endothelial cells by increasing nitric oxide production and suppressing NF-κB activation, partly at least through AMPK activation. These effects may explain some of the observed vasoprotective properties of liraglutide, as well as its beneficial effects on the cardiovascular system.
Similar content being viewed by others


Introduction
Glucagon-like peptide-1 (GLP-1), a member of the proglucagon-derived peptide family, is a 30-amino acid gut hormone. It is secreted in a nutrient-dependent manner, stimulating insulin secretion and inhibiting both glucagon secretion and gastric emptying, thereby reducing postprandial hyperglycaemia [1, 2]. GLP-1 has been shown to have favourable cardiovascular effects in preclinical and clinical studies. Thus it has been reported that GLP-1 has cardioprotective and vasodilatory effects in mice [3]. The same group also reported that a GLP-1 receptor agonist called liraglutide activates cytoprotective pathways and improves outcomes after experimental myocardial infarction in mice [4]. A beneficial vascular effect of GLP-1 has also been reported in humans with type 2 diabetes and stable coronary artery disease [5]. The mechanisms through which GLP-1 influences cardiovascular function are complex and incompletely understood. We therefore investigated whether the GLP-1 analogue, liraglutide, which is an acylated GLP-1, has protective effects on vascular endothelial cells. We also investigated the potential mechanism of its action.
Methods
Cell culture
Human umbilical vein endothelial cells were obtained from Clonetics (San Diego, CA, USA) and cultured in EGM2 medium supplemented with 2% (vol./vol.) fetal calf serum in the standard fashion. The cells in this experiment were used within three to four passages and were examined to ensure that they demonstrated the specific characteristics of endothelial cells. SV40 transformed endothelial cells (SVEC4) (murine endothelial cell line; ATCC, Rockville, MD, USA [6]) were also cultured in DMEM containing 10% (vol./vol.) fetal calf serum and used to measure nuclear factor κB (NF-κB) activation.
Western blot analysis
HUVECs were lysed using cell lysis buffer (Cell Signaling Technology, Beverly, MA, USA) containing 1 mmol/l phenylmethyl sulfonyl fluoride. Samples of HUVEC lysate were resolved on SDS-PAGE according to a standard protocol. After being transferred to membranes, the samples were immunoblotted with primary antibodies, followed by secondary antibodies conjugated to horseradish peroxidase. Bands were revealed using an enzyme-linked chemiluminescence detection kit (Amersham Biosciences, Piscataway, NJ, USA) and band density was quantified using an analyser (LumiVision; Aisin, Kariya, Japan). The following primary antibodies were used: anti-phospho-Ser-1177 endothelial nitric oxide synthase (eNOS) antibody, anti-AMP-activated protein kinase (AMPK) antibody, anti-phospho-Thr-172 AMPK antibody, anti-IκBα antibody, anti-phospho-IκBα antibody (all from Cell Signaling) and anti-eNOS monoclonal antibody (BD Biosciences, San Jose, CA, USA).
NF-κB activation
To study NF-κB activation, SVEC4 cells were stably transfected with a cis-reporter plasmid containing the luciferase reporter gene linked to five repeats of NF-κB binding sites (pNFκB-Luc; Stratagene, La Jolla, CA, USA), as previously described [7]. For this, the pNFκB-Luc plasmid was transfected together with a pSV2neo helper plasmid (Clontech, Palo Alto, CA, USA) into SVEC4 cells using a transfection reagent (FuGEN 6; Boehringer Mannheim, Mannheim, Germany). The cells were then cultured in the presence of G418 (Clontech) at a concentration of 500 μg/ml and the medium replaced every 2 to 3 days. Approximately 3 weeks after transfection, G418-resistant clones were isolated using a cloning cylinder and analysed individually for expression of luciferase activity. Several clones were also selected for analysis of NF-κB activation. Luciferase activity was measured using a luciferase assay kit (Stratagene).
We also measured changes in the levels of NF-κB p50 and p65 in nuclear extracts from HUVECs using a transcription factor assay kit (Active Motif Japan, Tokyo, Japan). Nuclear extracts were prepared with a nuclear extraction reagent (NE-PER; Pierce, Rockford, IL, USA), after which p50 and p65 were quantified using recombinant NF-κB p50 and p65 protein (Active Motif) as the standard.
Real-time PCR of HUVECs mRNA
For quantitative measurement of mRNA, 2 μg of total RNA was treated with DNase I for 15 min and subsequently used for cDNA synthesis. Reverse transcription was performed using a pre-amplification system (SuperScript; Gibco BRL, Gaithersburg, MD, USA) with random oligonucleotide primers. For details of the primer sequences see Electronic supplementary material (ESM) Table 1. A typical reaction (50 μl) contained 1/50 of RT-generated cDNA and 200 nmol/l of primer in 1× SYBR Green RealTime Master Mix (Toyobo, Tokyo, Japan) buffer. The PCR reactions were carried out in a LineGene system (BioFlux, Tokyo, Japan) under the following conditions: 95°C for 5 min, followed by 40 cycles at 95°C for 15 s, 60°C for 15 s and 72°C for 30 s.
Statistical analysis
Data are presented as the mean ± SEM. Multiple comparisons were evaluated by ANOVA followed by Fisher's protected least significant difference test. A value of p < 0.05 was considered statistically significant.
Results
Liraglutide increases nitric oxide production in HUVECs
Incubation of HUVECs with liraglutide (0.01 to 100 μg/ml) for 3 h increased the concentration of bioactive nitric oxide in the supernatant fraction of the cells (as measured by NO2 and NO3 levels) in a concentration-dependent manner (Fig. 1a). Examination of the time course for liraglutide (1 μg/ml) showed a substantial increase in nitric oxide production for 3 h, after which a modest elevation was observed (4–5 h) (Fig. 1b). eNOS activity, measured in terms of citrulline production, was observed to increase with liraglutide (1 μg/ml) treatment, which was attenuated by the AMPK inhibitor compound C (Fig. 1c).

Effect of liraglutide on nitric oxide (NO) production in HUVECs. a Concentration dependency of liraglutide: HUVECs were incubated with different concentrations of liraglutide for 3 h. b The time course of NO production induced by liraglutide: HUVECs were incubated with 1 μg/ml liraglutide for the indicated time periods. Nitric oxide in the medium was measured with an automated nitric oxide detector/HPLC system. Values are means ± SE (n = 6). c Effects of liraglutide on eNOS activity. HUVECs were incubated with liraglutide in the absence and presence of compound C (1 μmol/l), after which eNOS activity was assayed by measuring the formation of l-[3H]citrulline from l-[3H]arginine. Values are means ± SE (n = 3). *p < 0.05 compared with liraglutide; **p < 0.01 compared with basal
Liraglutide causes eNOS phosphorylation in HUVECs
eNOS phosphorylation was observed as early as 5 min after administration of liraglutide (1 μg/ml), reaching a maximum after 10 min in HUVECs. Following a similar time course, phosphorylation of AMPK and its downstream target, acetyl-CoA carboxylase was observed (Fig. 2a), but no Akt phosphorylation (data not shown). eNOS phosphorylation by liraglutide was attenuated by the AMPK inhibitor compound C, but not by the phosphoinositide-3-kinase (PI3K)/Akt inhibitor LY2294002 or the protein kinase A (PKA) inhibitor KT5720 (Fig. 2b). Liraglutide-induced phosphorylation of AMPK was not affected by co-treatment with an adenylate cyclase inhibitor, SQ 22536, or a cell-permeable cyclic AMP analogue (Fig. 2c).

a Liraglutide activates eNOS and AMPK in vascular endothelial cells. HUVECs were treated with liraglutide (1 μg/ml) for the indicated time periods before lysis, after which each cell lysate sample was probed with antibodies specific for phosphorylated (p) forms of eNOS, AMPK and acetyl-CoA carboxylase (ACC). Three independent experiments showed similar results. b eNOS phosphorylation by liraglutide (Lira) (1 μg/ml) was inhibited by compound C (CC 1 μmol/l), but not by LY294002 (LY 20 μmol/l) or KT-5720 (KT 500 nmol/l). c HUVECs were treated with liraglutide (1 μg/ml) alone or in the presence of adenylate cyclase inhibitor SQ 22536 (SQ 10 μmol/l) or a cell-permeable cAMP analogue pCTP-cAMP (cAMP 100 μmol/l). After 5 min of incubation, the cells were lysed and p-AMPK was analysed
Liraglutide preserves TNFα-induced downregulation of eNOS mRNA
Incubating HUVECs with TNFα markedly decreased eNOS mRNA levels, which was partially inhibited by co-treatment with an NF-κB inhibitor, BAY11-7082, which is known to selectively and irreversibly inhibit cytokine-induced IκB phosphorylation [8], suggesting that eNOS mRNA downregulation by TNFα may be at least partly NF-κB-dependent. Liraglutide attenuated the TNFα-mediated reduction in eNOS mRNA in a dose-dependent manner (Fig. 3).

Effect of liraglutide on TNFα-induced downregulation of eNOS mRNA. HUVECs were treated with TNFα (10 ng/ml) in the presence of different concentrations of liraglutide as shown for 16 h (Con, control: no TNFα), after which eNOS mRNA levels were measured using real-time PCR. Data represent the means ± SEM (n = 4) and are expressed as eNOS:GAPDH ratio
Liraglutide inhibits cytokine-induced NF-κB activation
We initially examined the effect of incubating liraglutide with TNFα for 2 h on NF-κB activation in SVEC4 cells. TNFα induced a sevenfold increase in NF-κB-mediated reporter gene expression. Liraglutide dose-dependently suppressed TNFα-elicited activation of NF-κB (Fig. 4a–e). We then examined the effects of compound C or AMPKα1 (also known as PRKAA1) small interfering RNA (siRNA) on liraglutide-induced inhibition of NF-κB activated by TNFα. Inhibition of NF-κB by liraglutide was significantly attenuated in compound C-treated cells (Fig. 4a) or siRNA-transfected cells (Fig. 4d,e). We also examined the effect of exendin(9–36), a classic GLP-1 receptor antagonist, on liraglutide-induced inhibition of NF-κB. This had no effect on inhibition of NF-κB, suggesting that the effect of liraglutide may have been mediated through a pathway independent of the GLP-1 receptor (Fig. 4f,g).

a–c Liraglutide inhibits TNFα-induced NF-κB-activation in SVEC4 cells. Liraglutide dose-dependently suppressed TNFα-activated NF-κB-dependent transcriptional activity, which was significantly attenuated in cells treated with compound C (1 μmol/l, a) or those transfected with AMPK siRNA (10 nmol/l, b), but not affected by exendin(9–36) (1 μmol/l, shown by the arrow, c). Squares, results in the absence of TNFα; white circles, results in the presence of TNFα only; black circles, results in the presence of TNFα plus additional treatment. Results are mean ± SEM (n = 6). **p < 0.01 vs NF-κB activity in the absence of liraglutide; † p < 0.05, †† p < 0.01 vs NF-κB activity in the presence of liraglutide. RLU, relative light units. d HUVECs were incubated with TNFα for 0 to 120 min. The cells were lysed and subjected to western blot analysis using anti-IκB-α and anti-phospho-IκB-α antibodies. e Effect of liraglutide on IκB-α degradation in HUVECs. Cells were incubated for 30 min with liraglutide (0.1 to 10.0 μg/ml), followed by TNFα for 15 min. Cells were then lysed and subjected to western blot analysis using anti-IκBα antibody. f, g HUVECs were stimulated with TNFα in the presence or absence of liraglutide (Lira1 1 μg/ml, Lira10 10 μg/ml) for 30 min, and NF-κB p65 (f) or p50 (g) subunits were quantified within nuclear extracts using a transcription factor assay kit. Results represent the mean ± SEM (n = 4). *p < 0.05, **p < 0.01
We then determined whether TNFα-induced NF-κB activation might occur through phosphorylation and subsequent degradation of IκB in HUVECs. To determine whether TNFα might induce IκBα phosphorylation, western blot analysis using antiphospho-Ser32 of the IκBα antibody was performed. TNFα was observed to induce IκB phosphorylation within 10 min, with reduced levels of phospho-IκBα being observed up to 60 min after probe (Fig. 4d,e). The blot was then re-probed with anti-IκB antibody, producing evidence of significant degradation within 10 to 30 min. After this, IκB synthesis was re-activated, possibly by NF-κB, at 60 min (Fig. 4d,e). Next, the effect of liraglutide on TNFα-induced IκBα degradation was determined 15 min after exposure to TNFα. Liraglutide dose-dependently inhibited TNFα-induced IκBα degradation, thereby suppressing NF-κB activation (Fig. 4d,e).
We also measured p50 and p65 in nuclear extracts from untreated HUVECs and in those extracts treated with TNFα in the presence and absence of liraglutide. Both p50 and p65 were markedly increased from very low levels by 30 min after stimulation with TNFα. This increase was significantly inhibited by liraglutide (Fig. 4f,g).
Liraglutide inhibits high glucose-elicited NF-κB activation
SVEC4 cells were incubated for 24 h with normal glucose (5.5 mmol/l) or high glucose (27.5 mmol/l), and were incubated for an additional 2 h in the absence and presence of TNFα to measure NF-κB activity. In each condition, the effect of liraglutide on NF-κB activation was examined. Incubation with high glucose caused higher NF-κB activity than with normal glucose, which was significantly inhibited by liraglutide. Similarly, high glucose enhanced TNFα-induced NF-κB activation in normal glucose, which was also attenuated by liraglutide (Fig. 5).

Cells were incubated for 24 h with normal glucose (Glu) (5.5 mmol/l) or high glucose (27.5 mmol/l), and then incubated for another 2 h in the absence and presence of TNFα to measure NF-κB activity. In each condition, the effect of liraglutide on NF-κB activation was examined. *p < 0.05 for high glucose vs normal glucose, †p < 0.05 for presence vs absence of liraglutide
Liraglutide inhibits VCAM1, ICAM1, E-selectin and MCP-1 mRNA induction in HUVECs
Incubation for 24 h with TNFα substantially induced expression of VCAM1, ICAM1, E-selectin and MCP-1. Induction of TNFα-induced gene expression was markedly suppressed by co-treatment with an NF-κB inhibitor, BAY11-7082 (not shown). Liraglutide (1 μg/ml) significantly inhibited TNFα-induced expression of these genes (Fig. 6a). Dose-dependent inhibition of NF-κB activation by aminoimidazole carboxamide ribonucleotide (AICAR) was observed when AICAR was added to cells 1 h before TNFα (Fig. 6b). AICAR at 1 mmol/l completely suppressed NF-κB activation. We next examined the effect of AICAR (0.5 mmol/l) on VCAM1, E-selectin, ICAM1 and MCP-1 mRNA levels. Those mRNA levels upregulated by TNFα were significantly attenuated by AICAR (Fig. 6c–g). However, GLP1R mRNA expression was not affected by AICAR treatment in the presence and absence of TNFα.

a Effects of liraglutide on TNFα-induced VCAM1, ICAM1, E-selectin and MCP-1 mRNA expression in HUVECs. Liraglutide (1 μg/ml) significantly inhibited VCAM1, ICAM1, E-selectin and MCP-1 mRNA. White bars, control; grey bars, control treated with liraglutide; black bars, TNFα alone; hatched bars, TNFα treated with liraglutide. Data are means ± SEM (n = 4) and expressed as a ratio of GAPDH. **p < 0.01 compared with the value of TNFα alone. b Dose-dependent suppression by AICAR of TNFα-induced NF-κB activation. White circles, no TNFα; black circles, TNFα-treated. c Effect of AICAR on VCAM1, d E-selectin, e ICAM1, f MCP-1 and g GLP-1R mRNA expression in the absence and presence of TNFα. **p < 0.01 compared with the value of TNFα in the absence of AICAR
Discussion
This study demonstrated that liraglutide induces nitric oxide production and inhibits TNFα-induced NF-κB activation in vascular endothelial cells. This conclusion is based on the finding that liraglutide caused eNOS phosphorylation and potentiated eNOS activity in HUVECs. Second, liraglutide inhibited TNFα-induced NF-κB activation and the NF-κB-dependent expression of various genes encoding inflammatory and cell adhesion molecules, including vascular cell adhesion molecule-1, E-selectin, intercellular adhesion molecule-1 and monocyte chemoattractant protein-1. This effect of liraglutide was also observed in the presence of high glucose. In addition, liraglutide restored cytokine-induced downregulation of eNOS mRNA. These data suggest that, in addition to lowering glucose levels, liraglutide might play a protective or anti-inflammatory role in the vessels of patients with type 2 diabetes.
We demonstrated AMPK activation by liraglutide in HUVECs and examined whether this might be associated with an increase in nitric oxide production. eNOS phosphorylation was seen to follow the same time course as AMPK activation. eNOS phosphorylation by liraglutide was attenuated by the AMPK inhibitor compound C, but not by the PI3K/Akt inhibitor LY2294002 or the PKA inhibitor KT5720. eNOS activation by liraglutide was also attenuated by compound C. Thus, AMPK activation by liraglutide could be an upstream effect of eNOS phosphorylation and activation. We also showed that liraglutide inhibits cytokine-induced NF-κB activation, which was restored by compound C or AMPK siRNA treatment. An AMPK activator, AICAR, was observed to suppress cytokine-induced NF-κB activation in vascular endothelial cells as well as in other type of cells [9, 10]. In the present study, we showed that AICAR suppresses cytokine-induced NF-κB activation, leading to inhibition of proinflammatory genes. However, AICAR had no effect on GLP-1R mRNA levels in the absence and presence of TNFα. These data suggest that AMPK activation by liraglutide might be responsible for inhibition of cytokine-induced NF-κB activation.
GLP-1 is widely believed to exert its actions through a distinct heptahelical G protein-coupled receptor (GLP-1 receptor), which is functionally associated with adenylate cyclase through the stimulatory Gs [11, 12]. GLP-1 may increase intracellular cyclic AMP (cAMP) and activate PKA, thereby increasing insulin secretion in beta cells. GLP-1 receptor has been shown to exist in HUVECs [13]. Thus, we examined whether cAMP might be associated with liraglutide-induced activation of AMPK. We found that phosphorylation of AMPK with liraglutide was not affected by co-treatment with an adenylate cyclase inhibitor SQ 22536 and that a cell-permeable cAMP analogue pCTP-cAMP did not cause AMPK phosphorylation and had no effect on liraglutide-induced phosphorylation of AMPK. GLP-1 is also thought to activate the PI3K/Akt pathway through epidermal growth factor receptor via GLP-1 receptor activation in beta cells [14]. However, the vascular effect of liraglutide was not altered by the PI3K/Akt inhibitor LY2294002. Thus, AMPK activation by liraglutide occurs independently of cAMP/PKA or PI3K/Akt to increase nitric oxide and protect against endothelial inflammation. Furthermore, exendin(9–36), a classic GLP-1 receptor antagonist, had no effect on liraglutide-induced inhibition of NF-κB [15]. This beneficial effect of liraglutide might therefore be mediated at least partly through a pathway independent of the GLP-1 receptor.
Two previous reports have implied that GLP-1 analogues might exert anti-inflammatory properties [16, 17]. One of them showed that exendin-4 inhibited interleukin-1β-induced inducible nitric oxide synthase at the protein level in RINm5F beta-cells [16]. The other report demonstrated that exendin-4 inhibits monocyte adhesion to endothelial cells and attenuates atherosclerotic lesions in apolipoprotein E-deficient mice, in which exendin-4 suppressed NF-κB activation in macrophages [17]. In both reports, the anti-inflammatory effect was reversed by cAMP inhibitor or a protein kinase A inhibitor, suggesting that this effect of GLP-1 analogues is mediated through the GLP-1 receptor. However, the present study suggests that the beneficial effect of liraglutide might be mediated through a pathway independent of the GLP-1 receptor. This difference might have been caused by the different cell type studied, but remains to be elucidated. It has been shown that GLP-1(9–36)amide, to which GLP-1(7–36)amide is rapidly metabolised by the enzyme dipeptidyl peptidase-4 (DPP-4), is a cyclic GMP-producing vasodilatory molecule capable of cardioprotective effects in hearts isolated from Glp1r knockout mice (Glp1r −/−) [3]. This implies the existence of an alternative receptor for GLP-1(9–36)amide. Furthermore, suppression of the cardioprotective and vasodilatory effects of GLP-1 by the DPP-4 inhibitor sitagliptin suggests that GLP-1(9–36)amide may function as a key intermediary in a subset of the cardiovascular effects of GLP-1 [3]. Thus, it is possible that the beneficial effect of liraglutide might be mediated through the possible receptor for GLP(9–36)amide, but this awaits clarification.
Native GLP-1 is a polypeptide that undergoes rapid metabolism by DPP-IV and, in addition, is rapidly cleared by renal elimination [18]. The half-life after intravenous or subcutaneous administration in humans has been reported to be ∼5 min [19]. Liraglutide has been reported to show an elimination half-life of 8.1 h [19]. This increase in the half-life of liraglutide is most likely to be mediated via a lower susceptibility to metabolism by DPP-IV [20] and a high degree of albumin binding of liraglutide (as has been shown for other fatty acid derivatives [21, 22]); moreover, after subcutaneous administration, an additional prolongation of the half-life is mediated by slow absorption of liraglutide from the injection site, as evidenced by the further increase in half-life observed with subcutaneous vs intravenous administration. Thus, liraglutide treatment probably induces much higher plasma levels compared with native GLP-1 concentration in plasma [19, 23, 24]. Therefore, the concentration of liraglutide required to produce beneficial effects in HUVECs (0.1 μg/ml = 26.6 nmol/l) might be achievable with the therapeutic doses used to treat patients with type 2 diabetes.
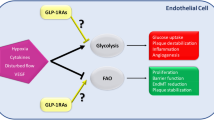
We provide a schematic diagram showing how liraglutide could suppress endothelial cell inflammation (see ESM Fig. 1). In conclusion, liraglutide exerts an anti-inflammatory effect on vascular endothelial cells through increased nitric oxide production and suppression of NF-κB activation, which is at least partly mediated via AMPK activation. These effects may explain some of the observed vasoprotective properties of liraglutide, as well as its beneficial effects on the cardiovascular system.
Abbreviations
- AICAR:
-
Aminoimidazole carboxamide ribonucleotide
- AMPK:
-
AMP-activated protein kinase
- cAMP:
-
Cyclic AMP
- DPP-4:
-
Dipeptidyl peptidase-4
- eNOS:
-
Endothelial nitric oxide synthase
- GLP-1:
-
Glucagon-like peptide-1
- NF-κB:
-
Nuclear factor κB
- PI3K:
-
Phosphoinositide-3-kinase
- PKA:
-
Protein kinase A
- pNFκB-Luc plasmid:
-
cis-Reporter plasmid containing the luciferase reporter gene linked to five repeats of NF-κB binding sites
- siRNA:
-
Small interfering RNA
- SVEC40:
-
SV40 transformed endothelial cells
References
Drucker DJ (2006) The biology of incretin hormones. Cell Metab 3:153–165
Irwin DM (2001) Molecular evolution of proglucagon. Regul Pept 98:1–12
Ban K, Noyan-Ashraf MH, Hoefer J, Bolz SS, Drucker DJ, Husain M (2008) Cardioprotective and vasodilatory actions of glucagon-like peptide 1 receptor are mediated through both glucagon-like peptide 1 receptor-dependent and -independent pathways. Circulation 117:2340–2350
Noyan-Ashraf MH, Momen MA, Ban K et al (2009) GLP-1R agonist liraglutide activates cytoprotective pathways and improves outcomes after experimental myocardial infarction in mice. Diabetes 58:975–983
Nyström T, Gutniak MK, Zhang Q et al (2004) Effects of glucagon-like peptide-1 on endothelial function in type 2 diabetes patients with stable coronary artery disease. Am J Physiol Endocrinol Metab 287:E1209–E1215
O’Connell KA, Edidin M (1990) A mouse lymphoid endothelial cell line immortalized by simian virus 40 binds lymphocytes and retains functional characteristics of normal endothelial cells. J Immunol 144:521–525
Hattori Y, Suzuki M, Hattori S, Kasai K (2002) Vascular smooth muscle cell activation by glycated albumin (Amadori adducts). Hypertension 39:22–28
Pierce JW, Schoenleber R, Jesmok G et al (1997) Novel inhibitors of cytokine-induced IkappaBalpha phosphorylation and endothelial cell adhesion molecule expression show anti-inflammatory effects in vivo. J Biol Chem 272:21096–21103
Giri S, Nath N, Smith B, Viollet B, Singh AK, Singh I (2004) 5-Aminoimidazole-4-carboxamide-1-beta-4-ribofuranoside inhibits proinflammatory response in glial cells: a possible role of AMP-activated protein kinase. J Neurosci 24:479–487
Hattori Y, Suzuki K, Hattori S, Kasai K (2006) Metformin inhibits cytokine-induced NF-kB activation via AMPK activation in vascular endothelial cells. Hypertension 47:1183–1188
Mayo KE, Miller LJ, Bataille D et al (2003) International Union of Pharmacology. XXXV. The glucagon receptor family. Pharmacol Rev 55:167–194
Thorens B (1992) Expression cloning of the pancreatic beta cell receptor for the gluco-incretin hormone glucagon-like peptide 1. Proc Natl Acad Sci U S A 89:8641–8645
Ishibashi Y, Matsui T, Takeuchi M, Yamagishi S (2010) Glucagon-like peptide-1 (GLP-1) inhibits advanced glycation end product (AGE)-induced up-regulation of VCAM-1 mRNA levels in endothelial cells by suppressing AGE receptor (RAGE) expression. Biochem Biophys Res Commun 391:1405–1408
Buteau J, Foisy S, Joly E, Prentki M (2003) Glucagon-like peptide 1 induces pancreatic beta-cell proliferation via transactivation of the epidermal growth factor receptor. Diabetes 52:124–132
Daniel EE, Anvari M, Fox-Threlkeld JE, McDonald TJ (2002) Local, exendin-(9–39)-insensitive, site of action of GLP-1 in canine ileum. Am J Physiol Gastrointest Liver Physiol 283:G595–G602
Kang JH, Chang SY, Jang HJ et al (2009) Exendin-4 inhibits interleukin-1beta-induced iNOS expression at the protein level, but not at the transcriptional and posttranscriptional levels, in RINm5F beta-cells. J Endocrinol 202:65–75
Arakawa M, Mita T, Azuma K et al (2010) Inhibition of monocyte adhesion to endothelial cells and attenuation of atherosclerotic lesion by a glucagon-like peptide-1 receptor agonist, exendin-4. Diabetes 59:1030–1037
Kiefer TJ, Habener JF (1999) The glucagon-like peptides. Endocr Rev 20:876–913
Elbrønd B, Jakobsen G, Larsen S et al (2002) Pharmacokinetics, pharmacodynamics, safety, and tolerability of a single-dose of NN2211, a long-acting glucagon-like peptide 1 derivative, in healthy male subjects. Diab Care 25:1398–1404
Madsen K, Knudsen LB, Agersoe H, Nielsen PF, Thøgersen H, Wilken M, Johansen NL (2007) Structure–activity and protraction relationship of long-acting glucagon-like peptide-1 derivatives: importance of fatty acid length, polarity, and bulkiness. J Med Chem 50:6126–6132
Kurtzhals P, Havelund S, Jonassen I (1995) Albumin-binding of insulins acylated with fatty acids: characterization of the ligand protein-interaction and correlation between binding-affinity and timing of the insulin effect in-vivo. Biochem J 312:725–731
Markussen J, Havelund S, Kurtzhals P et al (1996) Soluble fatty-acid acylated insulins bind to albumin and show protracted action in pigs. Diabetologia 39:281–288
Herman GA, Bergman A, Stevens C et al (2006) Effect of single oral doses of sitagliptin, a dipeptidyl peptidase-4 inhibitor, on incretin and plasma glucose levels after an oral glucose tolerance test in patients with type 2 diabetes. J Clin Endocrinol Metab 91:4612–4619
Damholt B, Golor G, Wierich W, Pedersen P, Ekblom M, Zdravkovic M (2006) An open-label, parallel group study investigating the effects of age and gender on the pharmacokinetics of the once-daily glucagon-like peptide-1 analogue liraglutide. J Clin Pharmacol 46:635–641
Acknowledgements
This study was supported in part by a grant from the Japan Private School Promotion Foundation. The authors are grateful to K. Manaka and K. Akimoto (Institute for Medical Science, Dokkyo University School of Medicine) for technical assistance.
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
This article has been retracted due to copyright violation.
The retraction note to this article can be found online at http://dx.doi.org/10.1007/s00125-011-2394-z.
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM Table 1
(PDF 19 kb)
ESM Fig. 1
A schematic diagram of how liraglutide suppresses endothelial cell inflammation. Liraglutide increases eNOS activity and inhibits NF-κB activation at least partly via AMPK activation, leading to nitric oxide (NO) production and NF-κB suppression. These reduce vascular inflammation caused by inflammatory cytokines and/or high glucose (PDF 19 kb)
About this article
Cite this article
Hattori, Y., Jojima, T., Tomizawa, A. et al. RETRACTED ARTICLE: A glucagon-like peptide-1 (GLP-1) analogue, liraglutide, upregulates nitric oxide production and exerts anti-inflammatory action in endothelial cells. Diabetologia 53, 2256–2263 (2010). https://doi.org/10.1007/s00125-010-1831-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-010-1831-8