Article Text

Abstract
Objective To evaluate changes in residual platelet reactivity (RPR) over time, and bleeding and ischaemic events rate using 5 vs 10 mg maintenance dose (MD) regimens of prasugrel 1 month after acute coronary syndrome (ACS).
Background The optimal level of RPR with prasugrel may change over time after an ACS.
Methods After 60 mg loading dose of prasugrel (T0) followed by 10 mg/day for 1 month, patients were randomised to receive prasugrel 10 mg/day (n=95, group A) or 5 mg/day MD (n=98, group B) up to 1 year. RPR was assessed at T0, 37 (T1) and 180 days (T2). The primary end point was Bleeding Academic Research Consortium (BARC) bleeding events ≥2 between 1 and 12 months, and the secondary composite end point was cardiac death, myocardial infarction, stroke and definite/probable stent thrombosis.
Results From T0 to T1, RPR significantly increased in both groups A and B and the increase was higher for group B (δ ADP 10 µmol: 13.8%±14.7% vs 23.5%±19.2%, p=0.001). At T2 a lower rate of high RPR patients were found in group A (2.6% vs13.3%; p=0.014). The BARC type ≥2 bleeding occurred in 12.6% of group A versus 4.1% of group B (OR 0.29, 95% CI 0.09 to 0.94) and secondary end point in 2.1% vs 1.0% (p=0.542), respectively, without stent thrombosis.
Conclusions RPR increases shifting from 60 mg loading dose to 10 mg/day prasugrel MD with a further increase of RPR reducing prasugrel MD to 5 mg 1 month after ACS. Clinical value of these pharmacodynamic findings should be proved in larger clinical trials.
Trial registration number NCT01790854.
- PHARMACOLOGY
- PERCUTANEOUS CORONARY INTERVENTION
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Key questions
What is already known about this subject?
Prasugrel 10 mg/day maintenance dose is recommended for 1 year in patients with acute coronary syndrome (ACS) treated with drug-eluting stent.
What does this study add?
Physicians should carefully evaluate patient's ischaemic and bleeding risks in determining which prasugrel maintenance dose might be more efficacious.
How might this impact on clinical practice?
The pharmacodynamic findings of Bleeding Events and Maintenance Dose of Prasugrel (BLESS) study might provide an important basis for the robust design of future trials focused on the optimisation of level of platelet inhibition in ACS.
Introduction
The Trial to Assess Improvement in Therapeutic Outcomes by Optimizing Platelet Inhibition with Prasugrel–Thrombolysis in Myocardial Infarction (TRITON–TIMI-38) study showed that in patients with acute coronary syndrome (ACS) undergoing percutaneous coronary intervention (PCI) the use of prasugrel translated into reduced ischaemic event rate but it was also associated with increased major bleeding rate compared with clopidogrel.1 Furthermore, the Food and Drug Administration (FDA) review of TRITON study2 highlighted that the risk–benefit appears to be greatest early in therapy with prasugrel, with fewer end points prevented per bleed as therapy is continued. In addition, the increase of residual platelet reactivity (RPR) after early period of ACS3 ,4 and the association between bleeding events and low RPR are well known.5Thus, the optimal level of platelet inhibition with prasugrel may change over time after an ACS. Therefore, switching from prasugrel 10 mg/day maintenance dose (MD) to 5 mg/day MD 1 month after the index event may be considered as able to reduce bleeding events. We sought to evaluate RPR change over time and the occurrence of bleeding and ischaemic events using reduced MD of prasugrel (5 vs 10 mg) in patients with ACS 1 month after drug-eluting stent (DES) implantation.
Materials and methods
The Bleeding Events and Maintenance Dose of Prasugrel (BLESS) trial was an open-label, randomised, single-centre study, designed to evaluate whether 1 month after PCI for ACS the shifting of prasugrel MD from 10 mg to 5 mg/day may reduce bleeding events. For this purpose, all patients with ACS underwent second-generation or third-generation DES implantation and received 60 mg loading dose (T0) of prasugrel followed by 10 mg/day for 1 month. Thereafter, patients confirmed to be adherent to prasugrel therapy were randomised to receive prasugrel 10 mg/day (group A) or 5 mg/day MD (group B) up to 1 year. All patients received 325 mg of aspirin followed by 100 mg/day for at least 1 year. RPR was assessed by light transmittance aggregometry at T0, 37 (T1, 7 days after the randomisation at 10 or 5 mg of prasugrel) and 180 days (T2).6 The inclusion criteria were all patients with ACS (≤75 years) treated with PCI and dual antiplatelet therapy (aspirin plus prasugrel) at the Division of Cardiology of Careggi Hospital, Florence, Italy, and informed written consent. ACS included unstable angina with ST segment changes, non-ST segment elevation acute myocardial infarction (MI) and ST segment elevation acute MI.6 Full exclusion criteria included age <18 years, active bleeding, bleeding diathesis, coagulopathy; history of gastrointestinal or genitourinary bleeding <2 months; major surgery in the past 6 weeks; history of intracranial bleeding or structural abnormalities; suspected aortic dissection; any previous transient ischaemic attack/stroke; administration in the week before the index event of ticlopidine, clopidogrel, prasugrel, ticagrelor, thrombolytics, bivalirudin, low molecular weight heparin or fondaparinux; known relevant haematological deviations: haemoglobin <10 g/dL, thrombocytopaenia <100×109/L; use of Coumadin derivatives within the past 7 days; chronic therapy with prasugrel or ticagrelor; known malignancies or other comorbid conditions with life expectancy <1 year; known severe liver disease, severe renal failure; known allergy to the study medications; pregnancy. The study was supported by the investigators (ClinicalTrial.gov identifier: NCT01790854). The protocol was approved by local investigational review boards and performed in compliance with good clinical practice and the Declaration of Helsinki. All patients provided signed informed consent prior to any study procedure.
The safety primary end point was the occurrence of bleeding type ≥2 events according to the Bleeding Academic Research Consortium (BARC) criteria,7 during the randomised treatment period (from 1 to 12 months). The secondary composite exploratory end point was cardiac death, MI or stroke.6 Other secondary end points of the study were: (1) the occurrence of definite or probable stent thrombosis from 1 to 12 months (the angiographic follow-up was strongly encouraged);8 (2) the pharmacodynamic response in patients with MD prasugrel 10 mg/day compared with those who were randomised to MD prasugrel 5 mg/day; and (3) the incidence of high RPR patients defined as ADP 10 µmol ≥70%.
In-hospital adverse events were recorded before discharge. One-month, 6-month and 12-month clinical follow-up data were obtained in outpatient consultation. Adverse clinical events were independently adjudicated by an external clinical event committee whose members were unaware of the group assignments. All source documents concerning events were provided to the clinical event committee, for accuracy and completeness.
Discrete data were summarised as frequencies, whereas continuous data as mean±SD or median. The χ2 test or Fisher's exact test were used for comparison of categorical variables, and the unpaired two-tailed Student's t-test or Kolmogorov-Smirnov non-parametric test were used to test differences among continuous variables for the end point. Odds risk (OR) and 95% CI were calculated. The time course of changes of RPR between and within groups was made by analysis of variance (ANOVA). BARC bleeding event-free survival was estimated by the Kaplan-Meier method. The log-rank test was used to compare BARC type ≥2 bleeding between patients treated with prasugrel 5 mg/day MD and patients treated with prasugrel 10 mg/day MD. All tests were two-sided and a p<0.05 was considered significant. All analyses were performed using the software package SPSS V.19.0 (SPSS, Chicago, Illinois, USA).
The study was designed on the basis of the superiority principle. We hypothesised that the primary safety end point (BARC type ≥2) would be 8% for 5 mg prasugrel MD and 16% for 10 mg prasugrel MD, which corresponds to a 50% bleeding reduction with 5 mg MD of prasugrel. The planned enrolment of 450 patients with ACS (225 patients in arm 5 mg/day MD; 225 patients in arm 10 mg/day MD) provides 80% power for detecting this bleeding reduction at α-level of 0.05. However, since the interim analysis, planned 2-year after the start of the study, showed fewer than expected events, after a discussion within the steering committee, we decided to stop the study considering the need of a larger simple size and a longer enrolment period to achieve the objective of the BLESS study, other than a reduced resource available.
Results
Owing to fewer than expected events, the trial was prematurely stopped after enrolling 193 of 450 planned patients. Thus, between November 2012 and April 2014, a total of 193 patients were enrolled in the BLESS study (study flow, figure 1): 95 patients randomised to prasugrel MD 10 mg/day (group A) and 98 to prasugrel MD 5 mg/day (group B). Baseline clinical characteristics of two arm patients were well matched (see table 1). The incidence of diabetes, female gender, chronic renal failure, previous PCI and coronary artery bypass graft and reduced left ventricular ejection fraction were not different between two group patients. Overall, 29% presented with acute MI. Moreover, 61% of patients showed multivessel coronary artery disease and 15% left main disease (table 2). All patients underwent PCI using second-generation and third-generation DES, with a mean of 1.5±0.7 vessel treated, and with a mean stent length of 31±21 mm. At hospital discharge, the medical therapy prescribed was similar between groups. The online supplementary table S1 shows the outcome before randomisation period.
Baseline characteristics and clinical presentation at hospital admission of study population
Angiographic, procedural characteristics and discharge therapy of study population

BLESS trial flow chart. ACS, acute coronary syndrome; BLESS, Bleeding Events and Maintenance Dose of Prasugrel; PCI, percutaneous coronary intervention.
supplementary tables
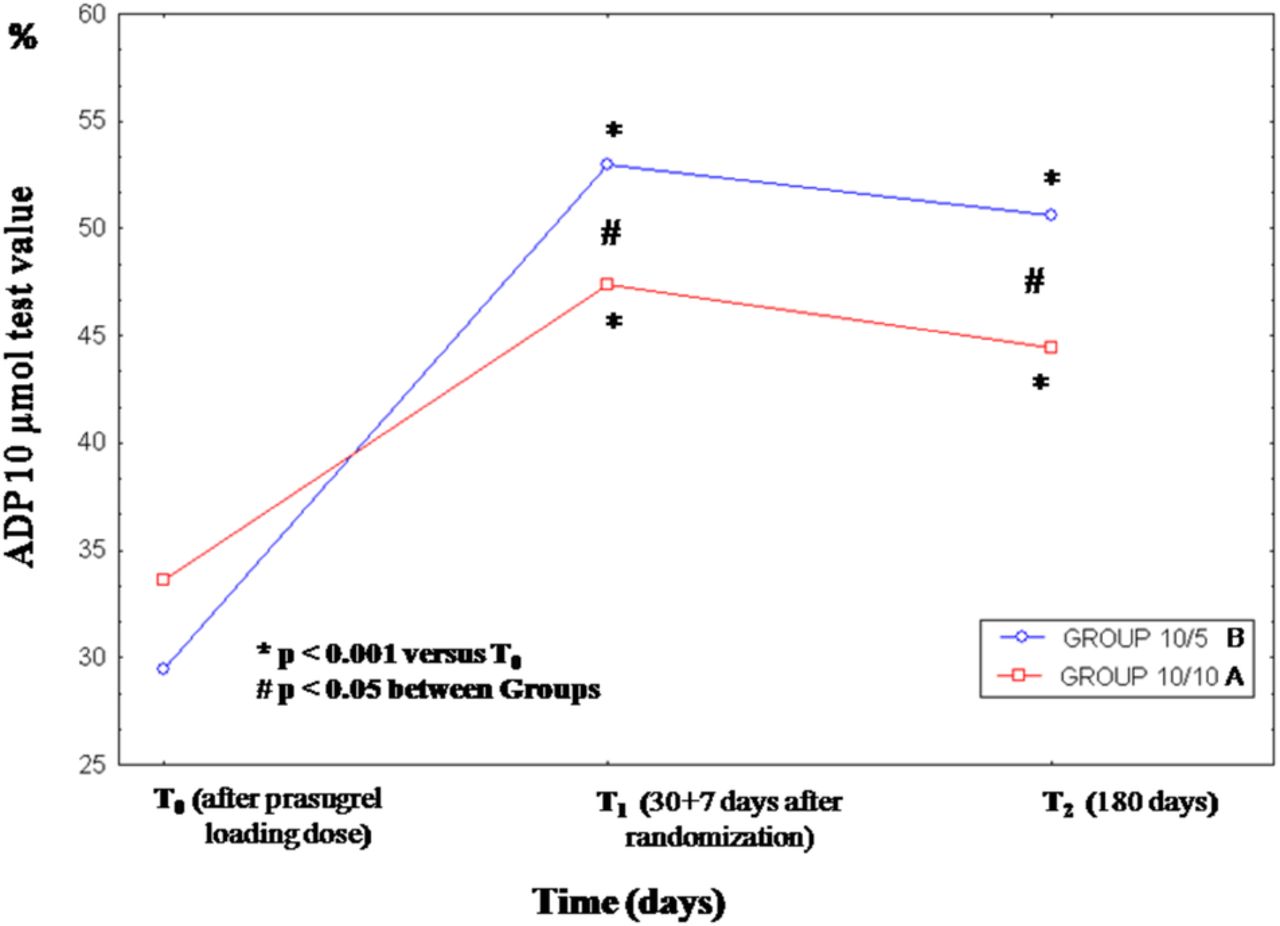
Out of 193 patients, RPR was assessed in 152, since 41 patients receiving IIb/IIIa inhibitors during the PCI procedures were excluded. The RPR at T0 were similar between group A and B (ADP 10 µmol: 34.5±16.2 vs 31.5±17.2; p=0.697, respectively). Out of 152, 2 (1.3%) high RPR patients were found. From T0 to T1, the RPR significantly increased in group A as well as in group B and the increase was higher for group B (δ ADP 10 µmol: 13.8%±14.7% vs 23.5%±19.2%, p=0.005), but from T1 to T2 no further increase of RPR was found in both groups (see ANOVA in figure 2 and online supplementary table S2). At T2 a lower rate of high RPR patients were found in group A compared with the group B (2.6% vs13.3%; p=0.014; table 2).

Time course and magnitude of changes of RPR within group B prasugrel 10/5 mg/day (─○─) and group A prasugrel 10/10 mg/day (─□─) and between groups. RPR, residual platelet reactivity.
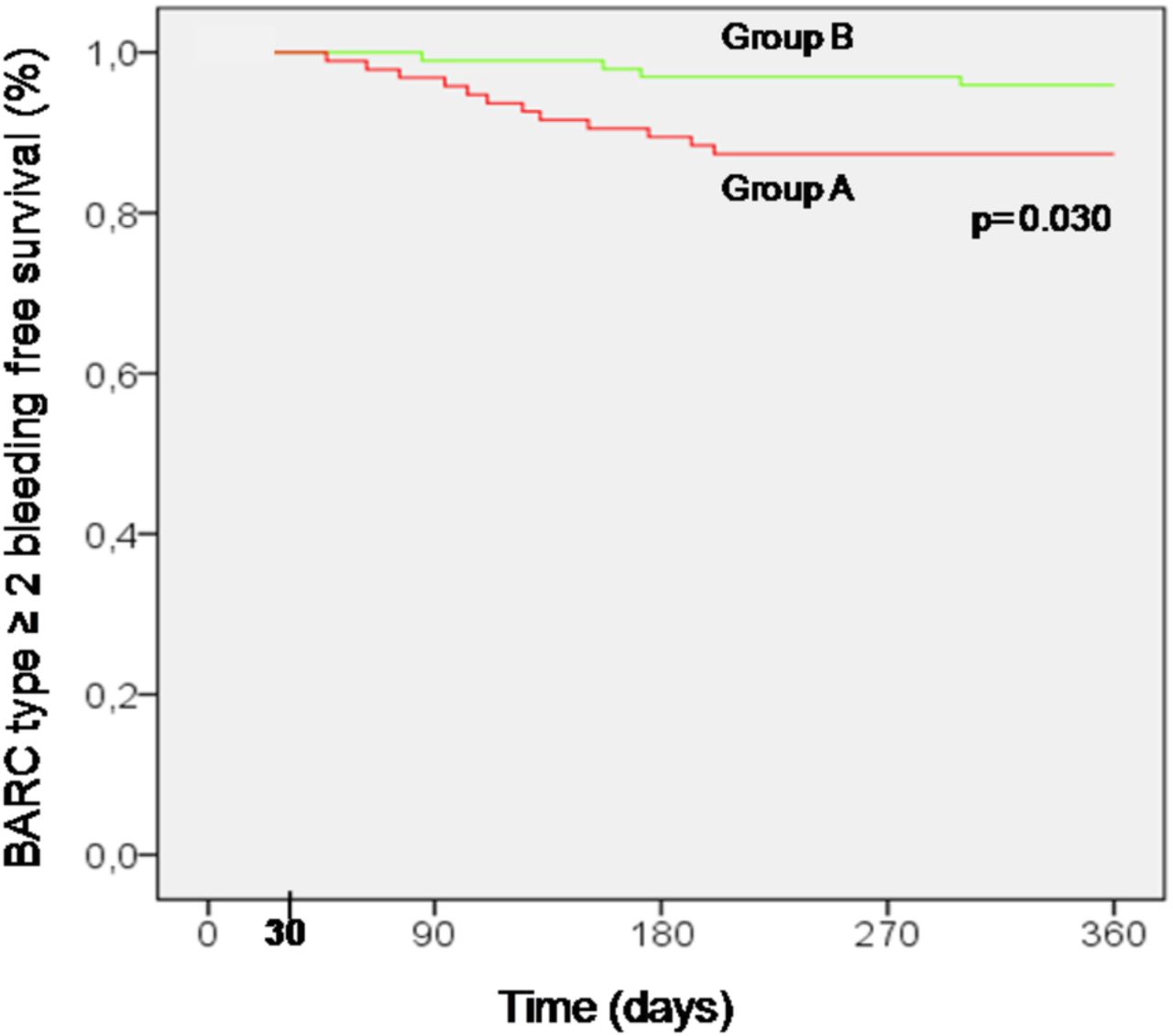
BARC bleedings were observed in 47.3% and 31.6% of group A and B (p=0.025); the BARC type 1 or 2 bleeding occurred in 45.3% vs 29.6% (p=0.024), the BARC type 2 in 10.5% and 2% (p=0.014), the BARC type 3a in 2.1% vs 2.0% (p=0.974), and the BARC type 2 or 3a in 12.6% and 4.1% (OR 0.29, 95% CI 0.09 to 0.94; p=0.031), respectively (see table 3). No BARC type 4 or 5 bleeding occurred. Event-free survival from BARC type ≥2 bleeding during randomisation period is shown in figure 3. Freedom from BARC type ≥2 bleeding events was higher in 5 mg/day prasugrel MD arm in comparison with 10 mg/day prasugrel MD arm (log-rank test, p=0.030).
Clinical outcomes during randomisation period (from 1 to 12 months)

{kind=link}
{kind=link}
{kind=link}
BARC type ≥2 bleeding-free survival according to the treatment with 5 mg/day prasugrel MD group B (─), or 10 mg/day prasugrel MD group A (─). Event rates were compared by log-rank test. BARC, Bleeding Academic Research Consortium; MD, maintenance dose.
The secondary composite exploratory end point occurred in 2.1% of group A (one MI, one stroke) versus 1.0% of group B (one MI), p=0.542 (see table 3). Considering the balance of safety and efficacy end point, the net clinical benefit was in favour of group B: 14.7% vs 5.1% (OR0.31, 95% CI 0.10 to 0.90, p=0.024). Urgent target vessel revascularisation rate was very low in both groups: 1 (1.1%) in group A versus 0 (0%) in group B (p=0.309). No definite or probable stent thrombosis occurred in both groups (clinical follow-up rate 100%, 6-month angiographic follow-up rate 83.9% (162/193)). During the randomisation period, two patients, both in group A, discontinued prasugrel: a gastric bleeding occurred at 6 months in the first patient and a haemorrhagic stroke occurred at 7 months in the second.
Discussion
A 5 mg/day prasugrel MD was used in the Targeted Platelet Inhibition to Clarify the Optimal Strategy to Medically Manage Acute Coronary Syndromes (TRILOGY ACS) trial9 to reduce bleeding complications in the vulnerable groups of elderly (>75 years) patients and younger, low body weight patients (<60 kg). RPR values of these patients using prasugrel MD 5 mg/day was non-inferior to prasugrel MD 10 mg/day used in younger, heavier patients.10 ,11 Moreover, the antiplatelet effects of the prasugrel MD 5 mg/day were greater than the effects with clopidogrel. These pharmacodynamic findings were confirmed in the larger TRILOGY platelet function substudy.12 Direct pharmacodynamic data to support the potential use of prasugrel MD 5 mg/day to reduce bleeding risk without an increase in the ischaemic risk in patients with ACS (≤75 years old) 1 month after 60 mg prasugrel loading dose (LD) for PCI are lacking. In the present study, we reported that the RPR increases between loading dose and 1 month, and an excess of RPR was found after shifting prasugrel MD from 10 to 5 mg/day. The concentration of active metabolite produced after 60 mg LD of prasugrel is higher (booster effect) in comparison to the one produced after 10 mg/day MD of prasugrel, explaining the change of RPR observed from T0 to T1 in this ACS population. In the present study, from T1 to T2 after ACS a trend towards a not significant reduction of RPR was observed, likely due to the spontaneous decrement of platelet reactivity. Thus, the combination of the lower production of active metabolite associated with MD prasugrel (favouring 5 vs 10 mg/day) and the spontaneous decrement of platelet reactivity after ACS may potentially explain the pattern of change of RPR observed in these patients. In addition, in the early phase of ACS, RPR values are influenced by several confounding factors, in particular by thrombus formation, the inflammation, the shear stress, the use of statin and other unknown variables. All these factors progressively either reduce their influence or disappear at 1 month.3 ,13 High on treatment RPR is well defined for clopidogrel and has been clearly identified as a risk marker for ischaemic events.14 High on treatment RPR can also exist with prasugrel in ACS after a LD of 60 mg and to a lower extent (<6% of treated patient) on the MD of 10 mg.15 Recent randomised trials as well as registry designed to evaluate whether more intensive antiplatelet therapy in acute phase, using high-dose clopidogrel16 ,6 or prasugrel,17 ,18 might improve clinical outcome failed to show the benefit of this strategy. Differently, the present study was designed to evaluate the possibility to optimise the level of platelet inhibition obtained with prasugrel after the acute phase of ACS, focusing on the concept that the optimal level of platelet inhibition may change overtime.
The adoption of the strategy reducing the MD of prasugrel from 10 to 5 mg/day might be able to optimise the risk/benefit profile of prasugrel in patients with ACS 1 month after second-generation or third-generation DES implantation. In fact, a 49% risk reduction in any BARC bleedings is associated with this strategy. Specifically, in the present study the most common bleedings are BARC type 1. However, nuisance-bleeding type 1 occurrence seems to be not significantly influenced by a reduced prasugrel MD strategy. On the contrary, an 83% risk reduction in BARC type 2 bleedings and, yet, a 71% risk reduction in BARC≥2 were associated with reduced prasugrel MD strategy. These findings are not negligible, since the occurrence of BARC type 2 bleedings, even minor, may be clinically relevant hurting the patient's day life and potentially reducing patient adherence to treatment with new antiplatelet agents such as prasugrel, favouring its disruption with potential consequences on ischaemic events.19 In fact, in the present study no discontinuation of prasugrel MD 5 mg was observed. Conversely, two patients taking 10 mg MD discontinued prasugrel.19 Finally, in the BLESS study the BARC≥2 bleeding event curves begin to separate soon after the randomisation period and continue to diverge throughout the follow-up favouring prasugrel MD 5 mg/day (figure 3), in keeping with previous and recent observations.20 ,21
Regarding the secondary exploratory efficacy end points we did not observe any increase in thrombotic events using prasugrel MD 5 mg/day. Surprisingly, we observed an impressive ‘0’ rate of definite or probable stent thromboses in the BLESS study population. Compared with bare metal stent and first-generation DES, the risk of definite or probable stent thrombosis is an average 50% lower with new generation DES.22 ,23 A significant reduction of stent thrombosis was observed in the TRITON1 and in the Platelet Inhibition and Patient Outcomes (PLATO)24 studies using first-generation DES in only 40% and 19% of cases, respectively. It is conceivable that our study benefited from the use of second-generation and third-generation DES. These devices were recently shown to be superior to the first-generation DES for revascularisation and clinical outcomes.25 This may be due to the fact that new generation DES with thin stent struts, advanced/absent polymers and improved antiproliferation agents along with better implantation techniques available, promote an early healing of stent struts favouring a near ‘0’ stent thrombosis. Finally, one should realise that in the early phase of an ACS, it is important to obtain and maintain an effective level of platelet inhibition. One month after an ACS, the optimal level of platelet inhibition may change and the adoption of a tailored strategy, using a reduced MD of prasugrel, might optimise the risk/benefit profile of prasugrel providing slightly less, but still consistent, platelet inhibition that translate into reducing bleeding risk without increasing the risk of thrombotic events. Of course, we recognise that no definitive conclusions can be drawn from the BLESS study; however, these findings can add some insights into the body of evidence of the dual antiplatelet therapy in ACS patient and may help us to customise the dual antiplatelet therapy according to the type of stent implanted in a patient with ACS.21 ,26
The BLESS study was prematurely interrupted resulting in the enrolment in a population underpowered for clinical events. Accordingly, the observed 71% risk reduction of BARC type ≥2 bleeding without any apparent increase of thrombotic events may be an overestimation of the magnitude of benefit and may be considered only as a hypothesis-generating finding. Moreover, whether the treatment benefit observed may be generalisable to non-thienopyridine P2Y12 inhibitor or other stent types24 ,27 ,28 is unknown. Despite these limitations, the pharmacodynamic findings associated with this strategy in current PCI patients have never been described and might provide an important basis for the robust design of future trials focused on the optimisation of level of platelet inhibition in patients with ACS.
Conclusions
The BLESS trial shows that in patients with ACS the RPR increases shifting from 60 mg loading dose to 10 mg/day prasugrel MD. A further increase in RPR and a higher rate of high RPR patients was observed shifting from 10 to 5 mg/day prasugrel MD after 1 month. Whether the adoption of this tailored strategy using a reduced MD of prasugrel might optimise the risk/benefit profile of this high-potency antiplatelet drug should be proved in larger clinical trials.
References
Footnotes
Contributors NC was involved in conception and design, drafting of the manuscript, final approval of the manuscript submitted. RM, RV, AMG, AM, VC and BB were involved in conception and design, analysis and interpretation of data. GP, RA, GFG and DA were involved in drafting of the manuscript or revising it critically for important intellectual content.
Funding The Bleeding Events and Maintenance Dose of Prasugrel (BLESS) trial was supported by the Arcard Onlus Foundation.
Competing interests GP reported receiving consulting fee from: AstraZeneca, Eli Lilly, The Medicines Company and Bayer. RM reported receiving honoraria for lectures from Daiichi Sankyo/Eli Lilly and Merck Sharp & Dohme. RA reported receiving consulting fees from Eli Lilly; lecture fees from Instrumentation Laboratory and Sigma Tau; and research grant funding from Bayer, BoehringerIngelheim and Pfizer. GFG reported receiving consulting fees from Bayer, BoehringerIngelheim and Eli Lilly; lecture fees from AstraZeneca, GlaxoSmithKline, Instrumentation Laboratory, Menarini and Sigma Tau; and research grant funding from Novo Nordisk, Merck Sharp & Dohme, Pfizer, Pierrel, Sanofi-Aventis and Servier. DA reported receiving consulting fees from Daiichi Sankyo/Eli Lilly and The Medicines Company and serving on the advisory boards of Cordis and CID.
Patient consent Obtained.
Ethics approval Careggi Hospital.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement No additional data are available.