Article Text

Abstract
Background Patients with pulmonary arterial hypertension (PAH) are managed according to evidence-based treatment guidelines.
Methods and results In this single-centre retrospective analysis, we examined outcomes of patients with PAH caused by congenital heart disease (PAH-CHD) with respect to exercise capacity and survival of adults treated with either bosentan or sildenafil monotherapy or bosentan-sildenafil dual therapy between January 2007 and January 2014. Of the 82 patients analysed, 29 had Down syndrome; 54 (65.8%) received bosentan monotherapy, 16 (19.5%) sildenafil monotherapy and 12 (14.6%) dual therapy. Mean treatment duration was 2.5 years for all patients and 4.1 years for 38 patients treated for ≥2 years. Pooled patient and treatment data showed initial improvement followed by stabilisation in mean 6 min walk distance (6MWD). For Down and non-Down patients, mean 6MWD increased and then stabilised on bosentan monotherapy. Mean 6MWD of patients on dual therapy at the time of analysis was 246.3 m before PAH-specific therapy initiation, 211.9 m immediately prior to addition of a second therapy and 214.4 m at last visit while on dual therapy. 1, 2 and 3-year survival rates for all patients from time of treatment initiation were 96%, 87% and 80%, respectively.
Conclusions For the majority of patients, monotherapy with a PAH-specific medication provided improved and sustained exercise benefits. For the small percentage of patients who required it, add-on therapy appeared to prevent further deterioration in exercise capacity but did not improve 6MWD.
- CONGENITAL HEART DISEASE
- Down syndrome
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Key questions
What is already known about this subject?
Long-term monotherapy with bosentan has been shown to result in sustained symptomatic benefits in patients with pulmonary arterial hypertension (PAH) caused by congenital heart disease (PAH-CHD), particularly in those without Down syndrome.
What does this study add?
This study expands on previous findings and demonstrates that in the proportion of patients with PAH-CHD who deteriorate on monotherapy, combination therapy can prevent further deterioration.
How might this impact on clinical practice?
For those patients who show a decline in exercise capacity after a period of stabilisation with monotherapy, addition of a second oral PAH-specific drug may help limit further deterioration.
Introduction
Up to 10% of patients born with heart defects that result in an incomplete separation of the systemic and pulmonary circulations develop the secondary condition of pulmonary arterial hypertension (PAH).1 ,2 The consequences of PAH associated with congenital heart defects (PAH-CHD) can be serious with the respective risk of mortality and morbidity being twofold and threefold higher in patients with PAH-CHD compared with those without PAH.3 Symptomatically, patients with PAH typically experience dyspnoea, fatigue, chest pain and occasionally presyncope on exertion. Such symptoms often interfere with everyday tasks that entail physical activity.4 Oral PAH therapies, such as sildenafil (a phosphodiesterase inhibitor) and bosentan (an endothelin receptor antagonist (ERA)) have been shown in clinical trials to improve exercise capacity in patients with PAH,5–7 including those with PAH-CHD.8 While the majority of patients with PAH are initially treated with monotherapy, newer guidelines recommend combining drugs from different classes in patients who fail to have an adequate response to initial therapy.9 ,10 Real-life data from registries or single-centre patient databases have shown that the use of combination therapy ranges from 46–75% in patients with idiopathic PAH,11 ,12 to 29–50% in patients with PAH associated with connective tissue disease13 ,14 and 17–32% in patients with PAH-CHD due to large unrepaired defects.15 In our centre, patients are treated in accordance with currently available evidence-based guidelines, with ERAs generally used as first-line therapy in patients with Eisenmenger's syndrome. Here, we conducted a retrospective single-centre analysis of adults with PAH-CHD who received bosentan or sildenafil monotherapy, or dual bosentan and sildenafil therapy. The aim was to examine outcomes in terms of symptomatic improvements based on exercise capacity and survival from treatment initiation.
Methods
Data on all patients (≥18 years old; with and without Down syndrome) with PAH-CHD who were referred to, and received PAH therapy, at the tertiary adult congenital cardiac centre at the Manchester Royal Infirmary, UK, were prospectively collected in a dedicated database. All data were collected as part of submission for the national UK database, which is mandatory for UK centres treating pulmonary hypertension patients. All non-Down syndrome patients, apart from one who refused to give consent, were diagnosed via right heart catetherisation (RHC), which confirmed the presence of severe PAH. Given the requirement to perform RHC under general anaesthesia in patients with Down syndrome, diagnosis for these patients was instead via echocardiography together with clinical signs and symptoms. Data included patient demographics, underlying cardiac diagnoses, WHO functional class, concomitant iron replacement therapy, initiation of PAH-specific medication, 6 min walk distance (6MWD), oxygen saturations (measured using pulse oximetry or via blood sampling) at rest and postwalk test, laboratory measurements of aspartate aminotransferase (AST), γ glutamyl transferase (GGT), haemoglobin and platelet levels, and all-cause mortality.
All patients treated with bosentan and/or sildenafil between January 2007 and January 2014 were included in this analysis. Routine vasoreactivity was not carried out unless it was felt that PAH was reversible. On the basis of clinical history and CHD diagnosis, most cases in this retrospective analysis were judged to be not reversible. Bosentan was initiated at 62.5 mg twice daily for the first 4 weeks and, tolerability permitting, titrated up to 125 mg twice daily thereafter. Sildenafil was initiated at a dose of 25 mg two to three times a day and uptitrated as clinically indicated and tolerated. Consistent with evidence-based guidelines, an ERA was generally chosen as first-line therapy. In the event of a lack of improvement in functional class (no change from class III to II or I) or if 6MWD fell by 20% from last visit then patients were switched from monotherapy to bosentan-sildenafil dual therapy.
All patients provided verbal consent to receive treatment as part of their routine care and hence consent was not formally documented. Patient data were collected by the adult congenital nurse specialist from the unit and were anonymised by the adult congenital nurse specialist and another study author. The adult congenital nurse specialist had access to patient identifying information but none of those who analysed the data had access to this information. As the lead clinician for the service, one of the study authors interacted with and treated the patients. This is a retrospective analysis of an existing database which collects routine clinical care data, and forms part of the national UK database, which is mandatory for UK centres treating pulmonary hypertension. As this was not a prospective study but a retrospective analysis of existing data, approval from an Institutional Review Board or ethics committee was not sought.
Data were analysed for all patients and by Down syndrome status, as well as according to treatment regimen, that is, monotherapy with either bosentan or sildenafil, or bosentan-sildenafil dual therapy following an initial period of monotherapy with either drug. Data were analysed at baseline (ie, prior to initiation of therapy), and visits 1, 2, 3, 4 and 5, which approximately corresponded to months 3, 6, 12, 18 and 24, and latest visit. Data were considered missing if a visit was missed. For patients on dual therapy at the time of data analysis and because of the variability of monotherapy and dual therapy treatment periods, data for 6MWD were analysed at baseline (ie, prior to initiation of monotherapy treatment), at the time point prior to dual therapy initiation and at the time point of last visit. Patients who received dual therapy following a period of monotherapy were not included in the monotherapy analysis.
Treatment duration was defined from the date of treatment initiation to date of last available visit and was also calculated for patients who had data post-month 24. Treatment duration for patients on monotherapy was from date of treatment initiation to date of last available visit. For patients on dual therapy at the time of data analysis, the initial monotherapy treatment duration was from the date of treatment initiation for first therapy to date of commencement of second therapy, while dual treatment duration was from the date of commencement of second therapy to date of last available visit.
Data were analysed using SAS 9.4 (SAS Institute, Cary, North Carolina, USA). Observed data only were included in the analyses and imputations for missing data were not made. Categorical data were described by frequency and percentage; continuous data were summarised by their means and SD, or median and range. Age between Down and non-Down patients was compared using a two-sample t test assuming unequal variances (Satterthwaite method). 6MWD at baseline by treatment group for patients by Down syndrome status was compared using a one-way analysis of variance (ANOVA), adjusting for treatment. A multivariate random effects repeated measures ANOVA model with random intercept and time terms (linear and, if applicable, quadratic) was used to estimate change in 6MWD from baseline for all patients and by Down syndrome status regardless of treatment regimen, and for patients on bosentan monotherapy by Down syndrome status. A similar model, adjusting for time as a linear term (random intercept and slope), was used to assess change in prewalk test oxygen saturations from baseline and change in the difference pre to postwalk test in oxygen saturation over time for all patients. For patients on dual therapy at the time of data analysis, difference in 6MWD at baseline prior to the initiation of monotherapy and last visit and the point prior to initiation of a second therapy and last visit were tested for using the paired t test. Significance was set at p<0.05. Kaplan-Meier plots provided an estimate of survival from time of treatment initiation to last available visit. Patients who survived were censored at the January 2014 cut-off date. Survival data from patients with Down syndrome in comparison to those without Down syndrome were not subject to a log-rank test.
Results
The database included 82 patients with PAH-CHD, of whom 29 had Down syndrome, who had received PAH-specific medication at our centre. Demographics and baseline characteristics are shown in table 1. Average age was 38.1±14.0 years and most (58.5%) patients were female. Patients with Down syndrome were significantly younger than those without Down syndrome (33.9±8.4 vs 40.3±15.9 years; p=0.02). The majority of patients had Eisenmenger syndrome (n=52; 63.4%), with ventricular septal defect (n=22; 26.8%) and atrioventricular septal defects (n=20; 24.4%) being the most common defects. All patients were in WHO functional class III prior to the start of treatment. As most patients had uncorrected defects that were considered unsuitable for repair and, additionally, were judged to have non-reversible PAH, vasoreactivity tests were not undertaken. Thus, it was not possible to measure vasoreactivity in these patients and assess its relationship to therapeutic response.
Demographics, baseline characteristics and on-treatment parameters for all patients and by Down syndrome status
Patients were treated with either bosentan (n=54) or sildenafil monotherapy (n=16), or bosentan-sildenafil dual therapy (n=12) after a period of either bosentan (n=10) or sildenafil (n=2) monotherapy. Mean treatment duration for all patients, regardless of treatment regimen, was 2.5 years and of longer duration in patients without Down syndrome than in those with Down syndrome, a difference that was most likely attributable to patients with Down syndrome being younger on average (table 1). For the 38 patients who were treated beyond month 24, the mean duration of therapy was 4.1 years. Durations for each treatment regimen for all patients and by Down status are shown in table 2.
Baseline 6MWD, treatment duration and concomitant iron replacement therapy by treatment regimen
Iron replacement therapy was prescribed in 11 patients (table 1) and administered concomitantly with either bosentan monotherapy or dual therapy (table 2). Administration of iron replacement therapy was similar in patients without (n=7; 13.2%) and with (n=4; 13.7%) Down syndrome (table 1).
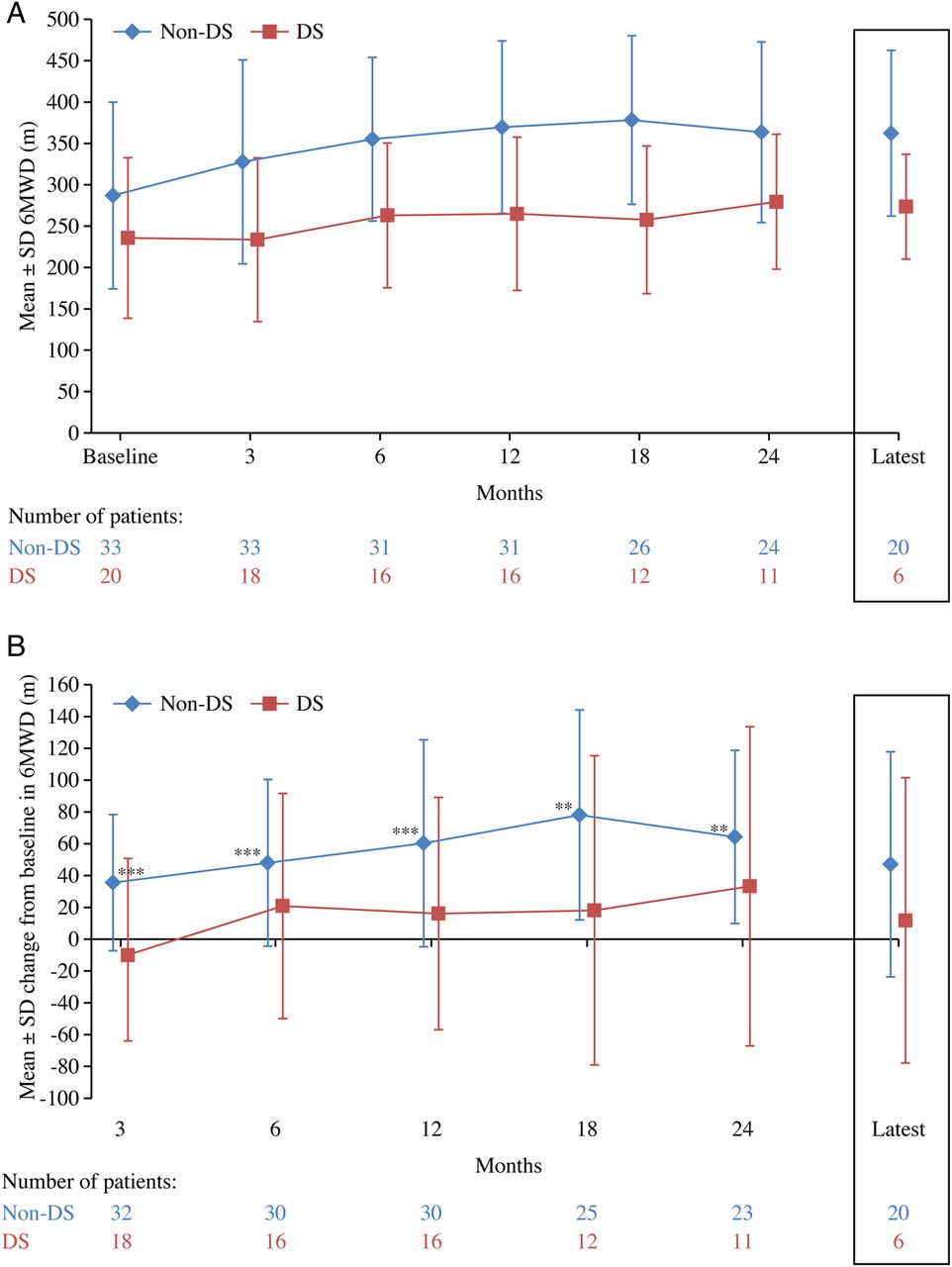
Patients without Down syndrome had a higher mean 6MWD at baseline (table 1) and at each time point (figure 1A) than those with Down syndrome. Following treatment, 6MWD tended to increase initially and then stabilise over time, regardless of Down syndrome status. In patients without Down syndrome the change in 6MWD from baseline was significant at time points up to month 24 and for those with Down syndrome changes from baseline were significant at months 12, 18 and 24 (figure 1B). 6MWD changed significantly over time for all patients when analysed together (p<0.01) and for patients without Down syndrome (p<0.01) but not for patients with Down syndrome (p>0.05).

(A) Absolute and (B) change from baseline in 6 min walk distance over time for all patients and by Down syndrome status. For the patients who had data post 24 months the mean±SD treatment duration from commencement of therapy to the latest time point was 4.1±1.1 (median 3.9 years, range 2.5–6.9 years) for all patients (n=38), 4.1±1.1 (median 3.8 years, range 2.5–6.1 years) for patients without Down syndrome (n=29) and 4.2±1.2 (median 4.0 years, range 2.6–6.9 years) for patients with Down syndrome (n=9). p Values tested whether change in 6MWD from baseline at each time point was significantly different from no change; *p<0.05, ***p<0.0001. 6MWD, 6 min walk distance; DS, Down syndrome.
There were no significant differences between 6MWD at baseline between treatment groups for patients without Down syndrome. For patients without and with Down syndrome, 6MWD initially increased and then stabilised over time with bosentan monotherapy (figure 2A). For patients without Down syndrome who were treated with bosentan monotherapy, the changes in 6MWD from baseline were significant at each time point up to month 24 (figure 2B). For patients with Down syndrome the change in 6MWD from baseline was not significant at any time point. 6MWD data for patients on sildenafil monotherapy are not described due to small patient numbers (n=2). It is important to note that assessment of response in patients with Down syndrome can be difficult to determine, as it is usually dependent on clinical symptoms and the views of those caring for the patient. Evidence that patients with Down syndrome respond less well to PAH-specific therapy may have more to do with the difficulty in assessing response than to any intrinsic difference in response to therapy compared with non-Down syndrome patients.
(A) Absolute and B) change from baseline in 6 min walk distance over time for patients on bosentan monotherapy by Down syndrome status. For the patients who had data post 24 months the mean±SD treatment duration from commencement of bosentan monotherapy to the latest time point was 4.1±1.1 (median 3.8 years, range 2.5–6.1 years) for patients without Down syndrome (n=21) and 4.5±1.5 years (median 4.4 years, range 2.6–6.9 years) for patients with Down syndrome (n=6). p Values tested whether change in 6MWD from baseline at each time point was significantly different from no change; **p<0.001, ***p<0.0001. 6MWD, 6 min walk distance; DS, Down syndrome.
Of the 10 patients without Down syndrome who were receiving dual therapy at the time of analysis, three were excluded because they did not have treatment-naïve baseline 6MWD data. For the seven patients with data, 6MWD at baseline was 246.3±73.0 m and 211.9±121.7 m at the visit prior to receiving dual therapy, representing a mean change of −34.4±124.3 m). 6MWD at last visit was 214.4±96.9 m, representing a mean change of −31.9±109.8 m from starting monotherapy (p=0.5) and of +2.6±33.0 m (p=0.8) from the visit prior to receiving dual therapy.
Mean pre and postwalk test oxygen saturations were similar between patients without and with Down syndrome (table 3). There was no statistically significant relationship between change in prewalk test oxygen saturation from baseline for all patients when analysed together or by Down syndrome status over time. The drop in oxygen saturation observed in the postwalk test at baseline was similar in magnitude to the drop at all time points. There was no evidence of a statistically significant difference between pre and postwalk test oxygen saturation over time for patients overall or by Down syndrome status.
Pre and postwalk test oxygen saturation and change in oxygen saturation over time for all patients and by Down syndrome status
With respect to laboratory parameters, mean AST levels were within the normal range at each time point, regardless of Down syndrome status (see online supplementary table S1). One patient had an elevated level of AST (267 U/L), although normal alanine aminotransferase level (42 U/L), at 18 months. There were no clinical consequences and levels of AST returned to and remained within normal levels for the 5 years of continued bosentan monotherapy. At all time points, mean GGT levels were lower for patients with Down syndrome compared with those without (see online supplementary table S1) but were within the normal range for both patient groups. Only patients on bosentan had sufficient GGT data available at each time point to analyse appropriately and levels were within the normal range at each time point.
Supplementary table
At all time points, mean haemoglobin levels were within the normal range regardless of Down syndrome status (see online supplementary table S1). Only patients on bosentan had sufficient haemoglobin data available at each time point to analyse appropriately, with haemoglobin levels within the normal range for these patients at each time point. Mean platelet counts were within the normal range (150–400×103/L) for patients without Down syndrome at all time points but were below normal range for patients with Down syndrome (see online supplementary table S1). This disparity was also reflected in patients on bosentan monotherapy.
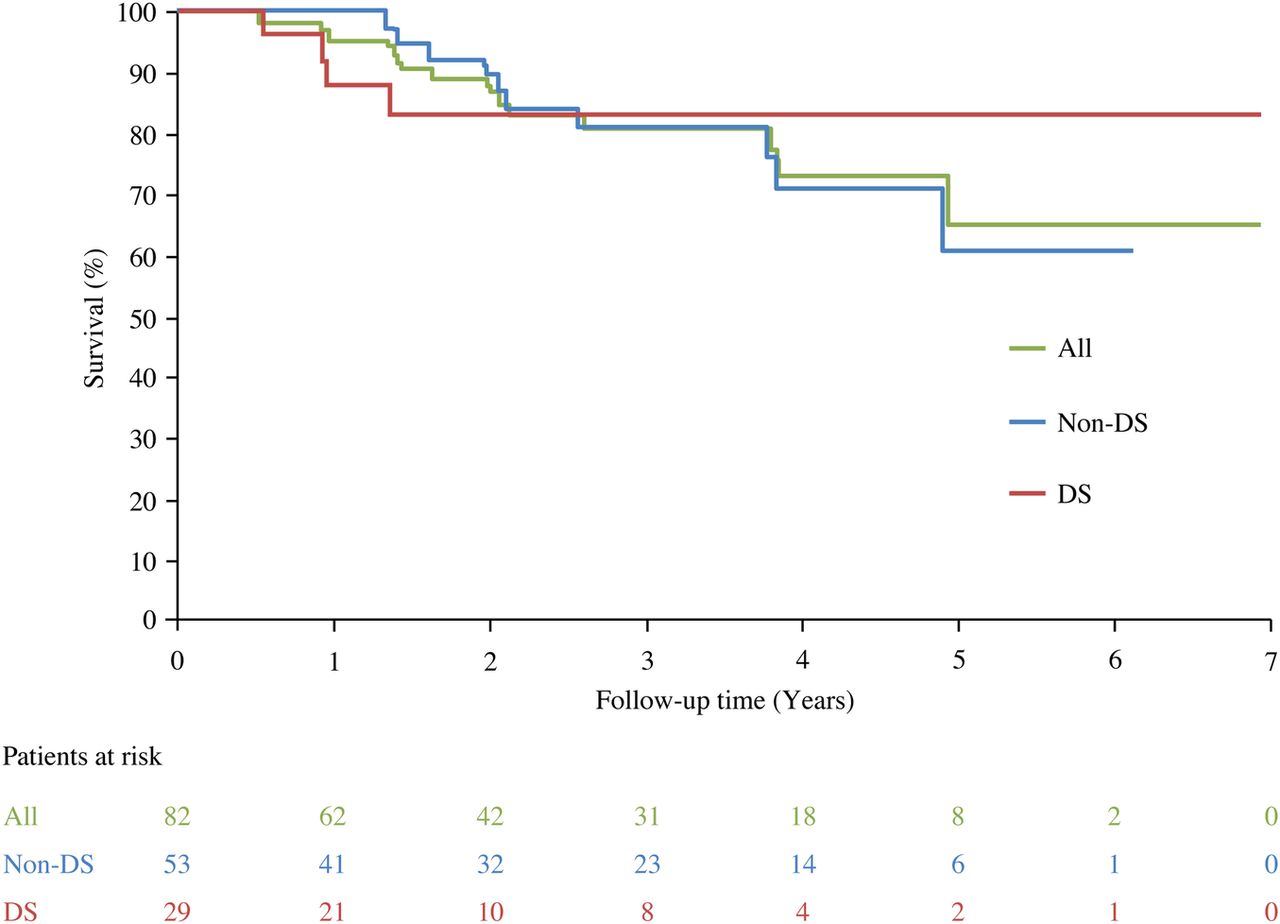
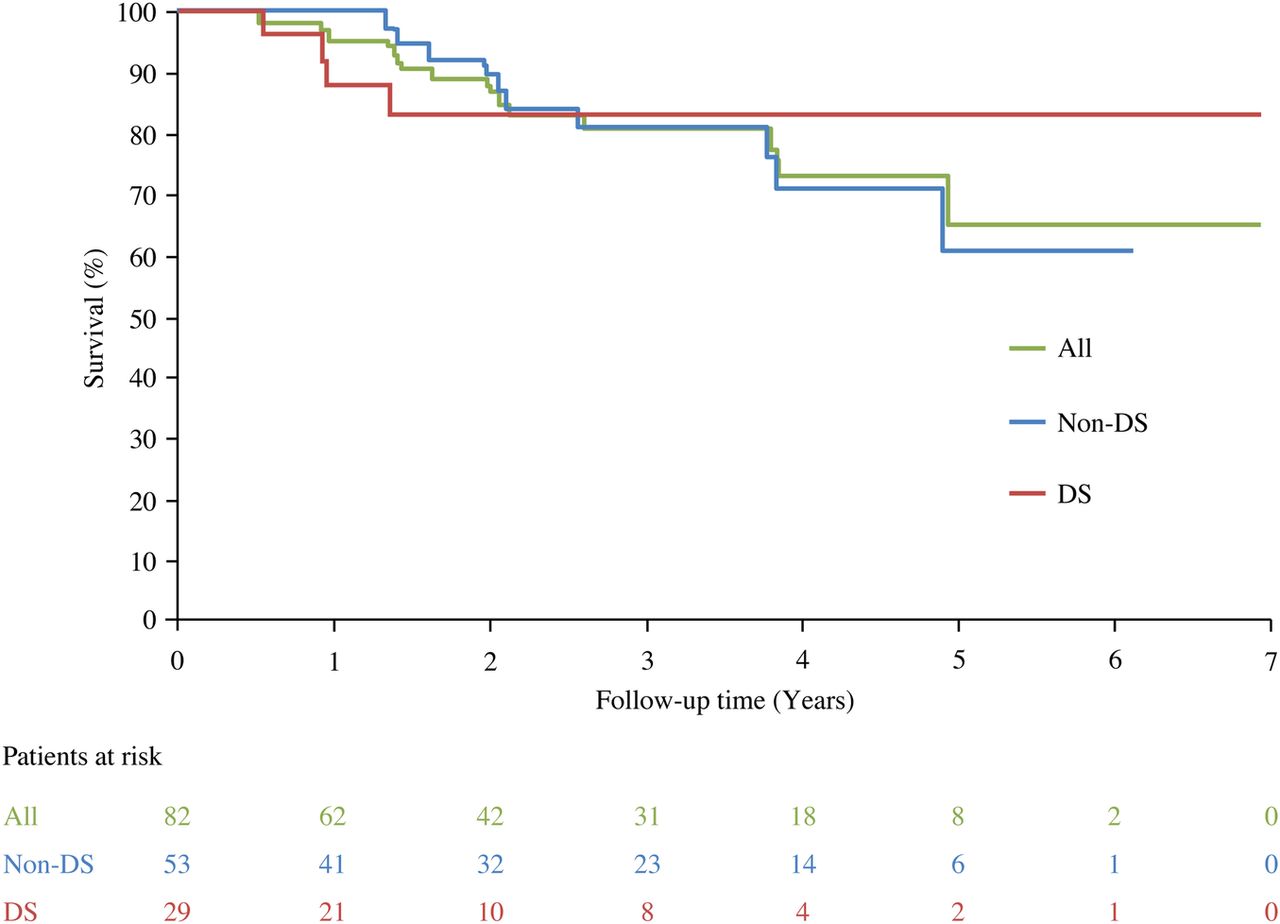
Over the follow-up period, 14 patients died, of whom four had Down syndrome. 1, 2 and 3-year survival rates for all patients from time of treatment initiation were 96%, 87% and 80%, respectively. For patients without Down syndrome the rates were 100%, 89% and 81%, respectively, and for those with Down syndrome they were 88%, 83% and 83%, respectively (figure 3). Cause of death varied and included sudden cardiac death, progressive right heart failure and death from thromboembolic complications.
{kind=link}
{kind=link}
{kind=link}
Survival from treatment initiation for all patients and by Down syndrome status. DS, Down syndrome.
Discussion
We have previously reported the outcomes of patients with PAH-CHD treated with bosentan monotherapy at our centre.16 The present analysis expands on these findings with outcomes from a larger number of patients, over a longer time period and with additional treatment regimens of sildenafil monotherapy and dual therapy with bostenan and sildenafil. Our findings show that, in patients with PAH-CHD, oral PAH-specific therapies significantly increase exercise capacity, measured by 6MWD, above baseline over the first 6 months of treatment with stabilisation up to 2 years. This is broadly consistent with other reports from the literature.17–21 Also consistent with other reports are the findings that patients with Down syndrome walk shorter distances at baseline and during treatment.18 ,20 We also observed a deterioration in exercise capacity beyond 2 years. Deterioration in 6MWD was not observed after a median follow-up of 3 years in a retrospective analysis in patients with Eisenmenger syndrome.21 However, 22% of patients in this study had treatment escalation (ie, increased dose or addition of a second therapy) after a median of 2.5 years.21 In our analysis, patients treated with dual therapy had a decrease in 6MWD from baseline preceding initiation of a second therapy, which although arresting deterioration in exercise capacity did not increase it to above predeterioration levels. This is in contrast to the findings of a recent prospective study, in which increases in 6MWD in patients with PAH and Eisenmenger syndrome were observed when sildenafil was added to bosentan in patients failing monotherapy.22 Our analysis, which is based on real-life observations rather than a rigorously controlled study, shows most patients do well on monotherapy while some require dual therapy and tend to have a modest response.
The drop in oxygen saturation from pre to postwalk test was consistent from baseline and at all time points. While treatment does not appear to increase postwalk test saturations it also does not substantially decrease them, thus supporting previous findings that PAH-specific therapies do not cause right-to-left shunting.8
Our analysis revealed no worrisome safety signals with regards to liver function and haematology during treatment. The observation of below normal platelet counts in patients with Down syndrome in our study is consistent with reports of abnormal haematology in this patient group.23 While platelet count is often abnormal in patients with Down syndrome, haemoglobin levels are usually in the normal range (13.0–18.0 g/dL in adult males and 11.5–16.5 g/dL in adult, non-pregnant females), although levels can also be lower and higher than normal.23 We observed no difference in haemoglobin levels between patients with and without Down syndrome, although mean values for all patients were slightly above the upper limits of the normal range, suggesting that in patients with PAH-CHD a higher value represents a normal mechanism for maintaining tissue oxygenation. It was not possible to measure the survival of patients from diagnosis because diagnosis was not always made at the time of therapy initiation in our centre. Rather, we measured survival over the treatment period. The estimated 3-year survival rates of 80% for all patients analysed in our study are in line with findings from the Registry to Evaluate Early and Long-Term PAH Disease Management (REVEAL) of 4-year survival estimates from enrolment of 77% in patients with Eisenmenger syndrome and of 76% in patients with PAH-CHD with unrepaired lesions.24 The 3-year estimates of 81% in patients without Down syndrome and of 83% in patients with Down syndrome are consistent with 4-year survival estimates from study entry of 72% and 80% in patients with PAH-CHD without and with Down syndrome.19
PAH is a rare condition and the diagnosis and management of patients tends to occur in designated pulmonary hypertension centres. Data from these centres on the disease course of the different PAH aetiologies and the most appropriate management and treatment strategies are often published and together with those from clinical drug trials feed into international10 ,25 and national management guidelines.26 Due to the complexity of an underlying cardiac defect, patients with PAH-CHD are often managed in centres able to provide specialist care in congenital heart disease and pulmonary hypertension. Our patients were treated in accordance with the most currently available management10 ,25 and commissioning27 guidelines. As such, the real-life data collected over a period of 7 years reflect the treatment and outcomes of patients within the recommended approaches that prevailed during this time. Bosentan monotherapy was the most frequently prescribed PAH-medication for patients without and with Down syndrome and prescribed for the longest time period. This approach is based on data from the BREATHE-5 trial of bosentan in Eisenmenger syndrome, which constitutes the earliest piece of rigorous evidence for the safety and efficacy of a PAH therapy in patients with PAH-CHD.8 Our database also included patients who received sildenafil monotherapy in more recent years. This reflects updated guidelines25 that give equal support to phosphodiesterase inhibitors and ERAs as first-line treatments for patients with PAH (in functional class III). Proportionally there are more patients with Down syndrome on sildenafil than patients without Down syndrome. This may be due to the necessity for frequent blood tests for liver function monitoring with ERAs, which might be difficult to obtain in some patients with Down syndrome.
The initiation of additional therapy is a relatively recent development at this centre and it has only been undertaken in a relatively small number of patients. PAH treatment guidelines currently give recommendations of when to initiate sequential combination therapy based on prognostic factors and treatment goals associated with an improved prognosis.25 However, in patients with PAH-CHD, prognostic factors such as right heart failure may not be as relevant as in idiopathic PAH. Moreover, the values set for treatment goals for 6MWD and cardiopulmonary and echocardiographic parameters are based on data from other PAH aetiologies and may not be the same for patients with PAH-CHD. While study data from the most experienced centres show combination therapy is used in patients with PAH-CHD,15 ,27 how widely it is used in less experienced centres is unknown as such centres rarely publish data. To encourage the uptake of dual therapy, more specific evidence and advice on when to initiate a second therapy in patients with PAH-CHD are required.
This is a retrospective analysis of data collected during routine clinical practice and is based on real-life observations. As such patients were not randomised and there were no matched controls. The principal limitation of our study is missing data at certain time points, either because patients missed a visit or because they had not been on treatment long enough to contribute to the time point analysed. Due to small patient numbers and missing data we were not able to rigorously analyse data from patients on sildenafil monotherapy.
In conclusion, our data show that in patients with PAH-CHD treated with oral PAH therapies in a specialised congenital heart disease centre, monotherapy with a PAH-specific medication seems to be effective in the vast majority of patients with PAH-CHD, and add-on therapy, while preventing decline, does not increase exercise tolerance. However, our data highlight the need for more evidence of when to initiate dual therapy in patients with PAH-CHD.
References
Footnotes
Contributors OM and VSM designed the study. OM, EH, LG, RC and VSM collected and analysed the patient data with additional statistical support. All authors critically reviewed the manuscript and approved the final version for submission.
Funding Actelion Pharmaceuticals Ltd provided funding to Elements Communications for medical writing and statistical analysis support. Medical writing support was provided by Lisa Thomas, PhD (Elements Communications Ltd, Westerham, Kent) and statistical support by Jonathan Alsop, PhD (Numerus Ltd, Wokingham, Berkshire).
Competing interests VSM has received research grants from Actelion Pharmaceuticals Ltd. RC reports honoraria from Actelion Pharmaceuticals Ltd, Bayer, GlaxoSmithKline, United Therapeutics and Eli Lily. LG received financial assistance from Actelion UK for producing a national patient's information leaflet on Eisenmenger syndrome in conjunction with the Somerville Foundation. OM and EH have no competing interests to declare.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement No additional data are available.