Article Text

Statistics from Altmetric.com
Vascular protective properties of supplemental glycine
Supplemental glycine, via activation of glycine-gated chloride channels that are expressed on a number of types of cells, including Kupffer cells, macrophages, lymphocytes, platelets, cardiomyocytes and endothelial cells, has been found to exert anti-inflammatory, immunomodulatory, cytoprotective, platelet-stabilising and antiangiogenic effects in rodent studies that may be of clinical relevance.1–17 The plasma concentration of glycine in normally nourished individuals—around 200 µM—is near the Km for activation of these channels, implying that the severalfold increases in plasma glycine achievable with practical supplementation can be expected to further activate these channels in vivo.18 ,19 The impact on membrane polarisation of such activation will hinge on the intracellular chloride content; cells which actively concentrate chloride against a gradient will be depolarised by channel activation, whereas other cells will experience hyperpolarisation. In cells that fail to concentrate chloride and that express voltage-activated calcium channels, glycine tends to suppress calcium influx; this effect is thought to mediate much of the protection afforded by glycine.1 The role of chloride channel activation in the mediation of glycine's physiological effects is commonly assessed by the concurrent application of the chloride channel inhibitor strychnine; if this abolishes glycine's effect, this effect is most likely mediated by chloride channels.
From the standpoint of vascular health, a recent report that glycine can stabilise platelets is of evident interest.7 When rats were fed diets containing 2.5–5% glycine, bleeding time approximately doubled, and the amplitude of platelet aggregation in whole blood triggered by ADP or collagen was halved. This effect was blocked by strychnine, and the investigators were able to confirm that platelets express glycine-gated chloride channels. They also demonstrated that human platelets likewise were glycine responsive and expressed such channels. Studies evaluating the interaction of glycine with aspirin or other pharmaceutical platelet-stabilising agents would clearly be appropriate, as would a clinical study examining the impact of supplemental glycine on platelet function.
Another recent study has established that cardiomyocytes express chloride channels.17 This may rationalise evidence that preadministration of glycine (500 mg/kg intraperitoneal) reduces the infarct size by 21% when rats are subsequently subjected to cardiac ischaemia-reperfusion injury; this effect was associated with increases in ventricular ejection fraction and fractional shortening in the glycine pretreated animals as compared with the controls.17 This protection was associated with a reduction in cardiomyocyte apoptosis, blunted activation of p38 MAP kinase and JNK and decreased Fas ligand expression. A previous study had reported that 3 mM glycine promoted increased survival of cardiomyocytes in vitro subjected to 1 h of ischaemia and then reoxygenated, and was also protective in an ex vivo model of cardiac ischaemia reperfusion.20
Vascular endothelial cells express glycine-gated chloride channels, and it has been suggested that glycine might exert an antiatherosclerotic effect by hyperpolarising the vascular endothelium.19 Since such cells do not express voltage-gated calcium channels, the impact of endothelial hyperpolarisation is to increase calcium influx, as calcium follows the charge gradient.21 This in turn would be expected to promote the calcium-mediated activation of endothelial nitric oxide synthase. Moreover, endothelial polarisation influences nicotinamide adenine dinucleotide phosphate (NADPH) oxidase activity; this is boosted by depolarisation and conversely inhibited by hyperpolarisation.22–24 The vascular-protective impact of potassium-rich diets is suspected to be mediated in part by the endothelial hyperpolarisation that results from modest physiological increases in the plasma potassium level (reflecting increased activity of the electrogenic sodium pump).25 ,26 Other factors being equal, an increase in endothelial nitric oxide generation coupled with a decrease in superoxide production could be expected to have an antiatherogenic and antihypertensive effect. Also speaking in favour of the antiatherosclerotic potential for glycine is a study demonstrating that glycine exerts an anti-inflammatory effect on human coronary arterial cells exposed to tumour necrosis factor (TNF) α in vitro; activation of NF-κB was suppressed, as was the expression of E-selectin and interleukin-6.27 So far, there are no published studies evaluating the impact of dietary glycine on atherogenesis in rodent models. Evidence that glycine has an antihypertensive effect in sucrose-fed rats is discussed below.
Glycine is a biosynthetic precursor for creatine, haeme, nucleic acids and the key intracellular antioxidant glutathione. Measures which raise or conserve intracellular glutathione levels may be of benefit from the standpoint of oxidant-mediated mechanisms that impair vascular health. A recent clinical study reports that concurrent supplementation of elderly participants with glycine and cysteine (100 mg/kg/day of each, cysteine administered as its N-acetyl derivative) reverses the marked age-related reduction in erythrocyte glutathione levels while lowering the serum markers of oxidative stress28; the authors, however, did not prove that the supplemental glycine was crucial for this effect.
With respect to diabetes, it is of interest that high intakes of glycine have the potential to oppose the formation of Amadori products, precursors to the advanced glycation endproducts (AGEs) that mediate diabetic complications.29 ,30 Indeed, supplementation of human diabetics with glycine—5 g, 3-4 times daily—is reported to decrease haemoglobin glycation.31 ,32 A similar effect has been reported in streptozotocin-treated diabetic rats.33 These studies did not measure AGEs per se, so their findings should be interpreted cautiously. Nonetheless, glycine supplementation has delayed the progression of cataract, inhibited microaneurysm formation, normalised the proliferative response of blood mononuclear cells and aided the humoral immune response in diabetic rats, effects which suggest that glycine may have potential for prevention of some diabetic complications.34–36 In a recent controlled but unblinded study, patients with diabetes experiencing auditory neuropathy achieved improvements in hearing acuity and auditory nerve conduction while ingesting 20 g glycine daily for 6 months.37
Glycine affords protection from sucrose-induced metabolic syndrome
Of particular interest are studies showing that high glycine intakes can counteract many of the adverse effects of a high-sucrose diet on the liver, adipose mass and vascular function in rats.38 ,39 Glycine decreased the elevated non-esterified fatty acid content of the liver of sucrose-fed rats, increased the state IV oxidation rate of hepatic mitochondria, corrected an elevation of blood pressure, normalised the serum triglycerides and insulin, prevented an increase in abdominal fat mass and, in the vasculature, boosted glutathione, decreased oxidative stress and normalised endothelium-dependent vasodilation. Of likely relevance to these findings is a recent clinical report that supplemental glycine (15 g daily in three divided doses) administered to patients with metabolic syndrome lessened indices of oxidative stress in erythrocytes and leucocytes, while lowering systolic blood pressure.40 These findings are of considerable interest, particularly in the light of evidence that high dietary fructose intakes can promote metabolic syndrome and non-alcoholic fatty liver disease in humans and increase LDL cholesterol.41–43
The protective effects of glycine in sucrose-fed rats, and in humans with metabolic syndrome, are not readily explained on the basis of the known metabolic effects of glycine. Fructose is known to exert its adverse effects primarily via its impact on liver metabolism; it is catabolised almost exclusively in the liver, and its oxidation, unlike that of glucose, is not regulated by metabolic need.41 ,44 As a result, a high intake of fructose floods the liver with substrate and suppresses hepatic fatty acid oxidation, while promoting de novo lipogenesis and triglyceride synthesis; increased generation of malonyl-coenzyme A is responsible for the first two effects, whereas an increase in glycerol-3-phosphate contributes importantly to fructose's stimulatory impact on triglyceride synthesis. These effects also increase hepatic production of diacylglycerols, which impair hepatic insulin sensitivity via activation of protein kinase C-ε.45 ,46 The increased triglyceride content of fructose-exposed hepatocytes can be expected to stabilise apoB100 and accelerate secretion of very-low-density lipoprotein (VLDL) particles47 ,48; this phenomenon may explain the elevation of LDL cholesterol induced by high-fructose intakes.42 The increased hepatic secretion of VLDL triglyceride presumably is responsible for the increase in visceral fat observed in rodents and humans fed high-fructose diets.41 This in turn can induce metabolic syndrome, including an increase in blood pressure driven in part by hyperinsulinaemia49 (figure 1).
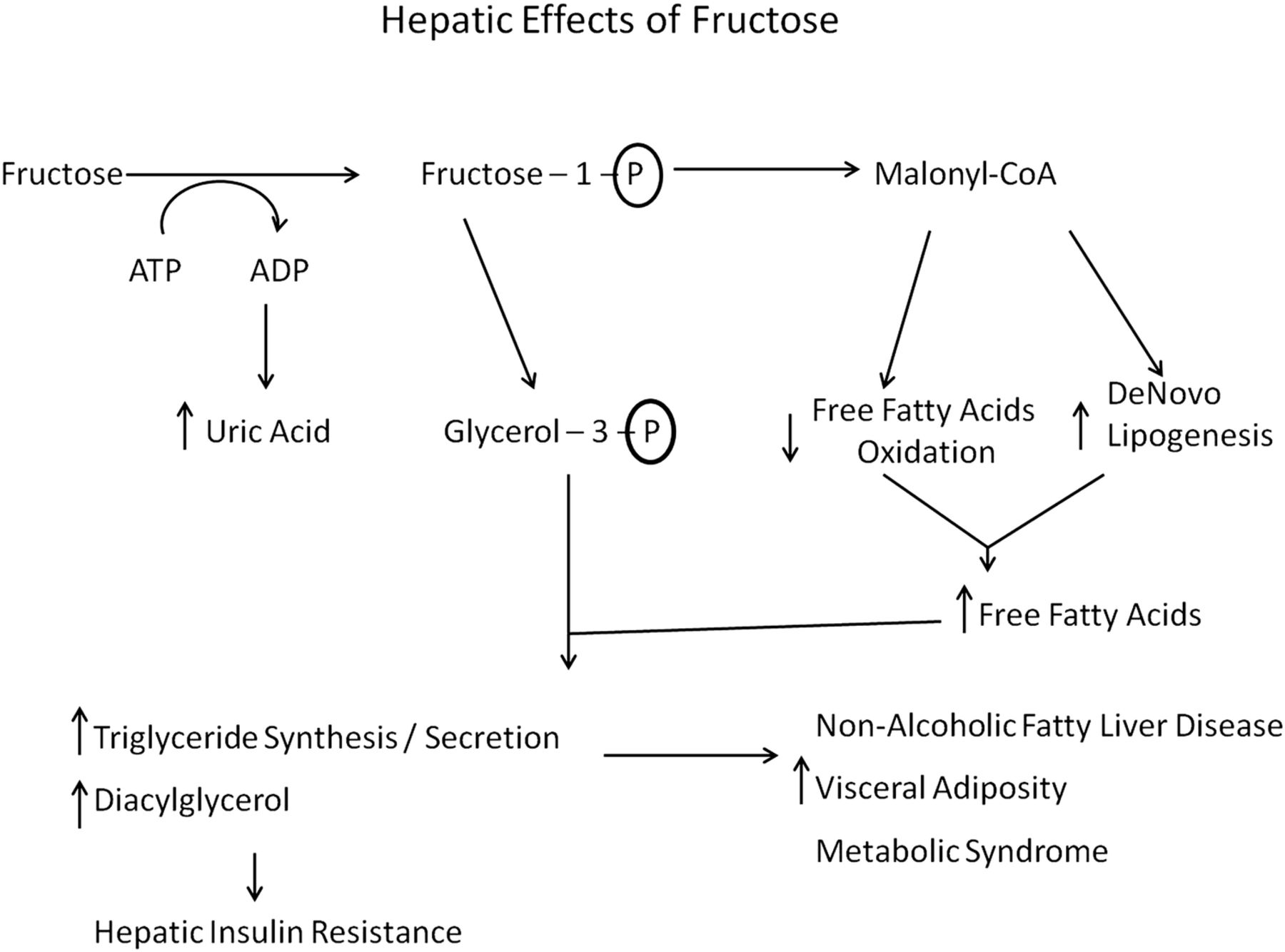
Hepatic effects of fructose.
There is recent evidence that fructose can also act indirectly to boost hepatic gluconeogenesis. Fructose, but not glucose, can activate AMP kinase (AMPK) in certain regions of the hypothalamus, resulting in increased adrenocortical production of corticosteroids that promote hepatic transcription of phosphoenolpyruvate carboxykinase, rate-limiting for gluconeogenesis.50
How does glycine intervene in this process? We propose that glycine-stimulated secretion of glucagon-like peptide-1 (GLP-1) and of glucagon itself plays a key role in this regard.
Glycine may stimulate GLP-1 and glucagon release
Gameiro et al,51 working with the GLUTag cell line derived from intestinal L-cells—the cell type specialised for GLP-1 production in the intestinal mucosa—,have found that glycine provokes an increase of GLP-1 secretion in these cells. This reflects an activation of glycine-gated chloride channels that triggers a reduction in membrane polarisation, leading to an increase in cytoplasmic free calcium and a consequent release of GLP-1. The ability of these chloride channels to decrease membrane polarisation in these cells reflects the fact that they concentrate chloride via a Na+-K+-2Cl− transporter. Drugs which inhibit either the glycine-gated channels or the chloride uptake mechanism prevent glycine from stimulating GLP-1 release in GLUTag cells. Since the apical microvilli of L-cells face the intestinal lumen, they are ideally positioned to detect an increase in glycine in the luminal contents. Hence, glycine supplementation could be expected to boost GLP-1 production. Although there do not appear to be any studies that have examined the GLP-1 response to orally administered glycine per se, there are two clinical studies demonstrating that plasma GLP-1 levels rise following ingestion of gelatin, a protein extraordinarily rich in glycine (constituting 30% of its amino acids).52 ,53 This may explain why, when glucose was fed to patients with type 2 diabetes in conjunction with seven different proteins, gelatin was second only to cottage cheese in potentiating the postprandial insulin response.54
Oral administration of glycine in humans (75 mg glycine/kg lean mass) has also been reported to stimulate an increase in glucagon secretion by pancreatic α-cells.55 This response is negated if glucose is ingested simultaneously, most likely reflecting the impact of glucose-evoked secretion of somatostatin from islet δ-cells.56 The contention that oral glycine stimulates GLP-1 production is difficult to square with glycine's impact on glucagon, as GLP-1 is known to inhibit α-cell glucagon secretion, either directly or by provoking δ-cell secretion of somatostatin.57 However, there is recent evidence that glycine may act directly on α-cells as a glucagon secretagogue—and perhaps this effect overrides that of GLP-1 (the impact of GLP-1 on somatostatin secretion might be minor when glucose is at basal levels, and that of GLP-1 receptor expression on α-cells is very low57). Li et al58 have shown that α-cells express glycine-gated chloride channels that, when activated, trigger an influx of calcium and glucagon release. This suggests that α-cells, like L-cells, have a mechanism for concentrating chloride intracellularly, such that a receptor-mediated increase in membrane permeability triggers chloride efflux and membrane depolarisation. Since the affinity of glycine-gated channels for glycine is close to the fasting concentration of glycine in plasma,51 it can be anticipated that a rise in plasma glycine induced via supplementation will cause an increase in glucagon secretion. One rather old study failed to observe an increase in glucagon secretion when glycine was infused intravenously, until the glycine reached supraphysiological levels59; it is not clear why the results of this study appear discordant with those of the two studies previously cited.
It is notable that GLP-1 and glucagon work in complementary ways to promote fatty acid oxidation and oppose lipogenesis in the liver.60–65 The effects of glucagon appear to be mediated primarily by cAMP, whereas GLP-1 triggers activation of AMPK in hepatocytes. Joint action of GLP-1 and glucagon on the liver could readily account for the ability of supplemental glycine to counteract the excessive hepatic triglyceride synthesis promoted by sucrose or fructose feeding (see figure 2). Indeed, GLP-1 agonists have been shown to protect against hepatic steatosis in sucrose-fed rats, and to have clinical utility in non-alcoholic fatty liver disease.66–70 Fortuitously, although glucagon could be expected to promote hepatic gluconeogenesis, GLP-1 mediated AMPK activation would tend to offset this effect.71 Indeed, AMPK suppresses the transcription of phosphoenolpyruvate carboxykinase in the liver, potentially offsetting the stimulatory impact of fructose-evoked cortisol in this regard.72–74

{kind=link}
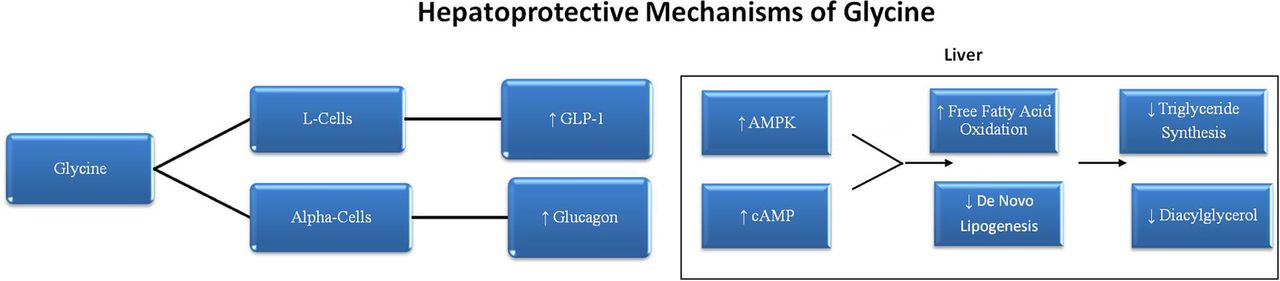{kind=link}
Hepatoprotective mechanisms of glycine.
Intriguingly, peptide drugs with dual agonism for GLP-1 and glucagon receptors have been developed recently, and these agents have shown markedly beneficial effects in mice with diet-induced obesity.75–77 They can induce a weight loss of 15–20%, modestly decrease calorie intake while boosting thermogenesis, decrease hepatic triglyceride levels and serum levels of triglycerides and LDL cholesterol, improve insulin sensitivity and glucose tolerance and counteract leptin resistance. These beneficial metabolic effects are only partially attributable to the associated weight loss and are considerably greater than the benefits seen with GLP-1 agonists alone. These agents provide continual stimulation of their target receptors, and hence understandably achieve more potent effects than dietary glycine, which at best could only boost GLP-1 and glucagon levels episodically. Nonetheless, while glycine does not induce weight loss or suppress calorie intake in sucrose-fed mice, it reduces visceral fat stores by over 50%, increases the thermogenic potential of hepatic mitochondria by increasing state 4 respiration, alleviates hepatic steatosis and improves insulin sensitivity and serum lipids. Hence, its effects are homologous to, if not as dramatic, as those seen with the coagonist drugs.
Further implications of GLP-1 upregulation
If supplemental glycine does indeed boost secretion of GLP-1, this may have interesting implications for the prevention and treatment of diabetes, and for the preservation of vascular health. As is well known, GLP-1 functions to potentiate glucose-stimulated insulin secretion, and this is the basis for the therapeutic utility in diabetes of analogues of GLP-1 such as liraglutide and exenatide which activate the GLP-1 receptor but have a vastly longer half-life owing to their resistance to degradation by dipeptidyl protease-4 (DDP-4).78 Endogenously produced GLP-1 has a half-life of only several minutes in plasma owing to its rapid degradation by DDP-4; hence, drugs which can safely inhibit DDP-4, such as sitagliptin, are currently employed to prolong the efficacy of endogenously produced GLP-1 in patients with diabetes.79 If supplemental glycine does indeed boost GLP-1 production, it presumably could be used as an adjuvant to DDP-4 therapy, and, as a stand-alone measure, might have some potential for the primary prevention of diabetes.
Moreover, if supplemental glycine can promote a physiologically meaningful increase in GLP-1 production, it may have broader protective potential than is currently appreciated, reflecting the diverse and largely protective physiological effects of GLP-1.80 With respect to vascular health, GLP-1 agonist drugs exert cardioprotective effects in rodent models of myocardial infarction and congestive failure.81–84 Clinically, they promote modest weight loss in patients with diabetes and obese non- diabeties, and exert favourable effects on systolic blood pressure, serum lipids, inflammatory markers and endothelial function.85 ,86 Readers interested in the vascular-protective properties of GLP-1 agonism can be referred to a recent review by Lorber.86 Assessing the impact of supplemental glycine on GLP-1 production should be a high clinical priority.
An impact on Kupffer cell activation
The marked utility of dietary glycine in rodent models of alcohol-induced steatosis has been traced to its ability to suppress Kupffer cell activation.9 ,87 Ethanol feeding, by promoting intestinal permeability, enables portal influx of bacterial endotoxins. The resulting activation of Kupffer cells exposes hepatocytes to proinflammatory cytokines such as TNF-α that play a key role in induction of steatohepatitis. Glycine antagonises Kupffer cell activation via glycine-gated channels, as previously discussed.
There is some recent evidence that high-fructose diets in rats likewise impair the intestinal barrier function, leading to an activation of Kupffer cells that exacerbates fructose-induced steatosis.88–90 It is reasonable to suspect that a high-glycine diet would be protective in this regard, as it is in rodent models of alcohol-induced steatosis. Whether Kupffer cell activation plays a role in the hepatic steatosis evoked by high-fructose diets in humans remains to be established.
Fortunately, glycine powder is inexpensive, highly soluble and has a pleasant sweet flavor; indeed, its name is derived from the Greek work for ‘sweet’.16 Clinically useful effects have been observed in patients with metabolic syndrome or diabetes with glycine intakes of 5 g, 3–4 times daily, without discernible side effects.32 ,40 Glycine is readily administered by blending into a fluid of choice, and it should lend itself well to incorporation into functional foods. Glycine intake can also be boosted by ingestion of gelatin.
Is uric acid a mediating risk factor?
The proposal that glycine might function as an ‘antidote’ to the adverse metabolic impact of fructose must contend with the fact that fructose can markedly amplify production of uric acid in the liver. Administration of a bolus of fructose leads to the rapid hepatic generation of ADP owing to the unregulated activity of fructokinase; this in turn can lead to accelerated production of AMP, adenosine and purine catabolites—including the ultimate catabolite (in humans) uric acid. The ability of fructose-rich diets to boost serum urate levels is well known, and there is no reason to suppose that glycine would prevent this effect. This urate does not pose a problem in fructose-fed rodents, as their uricase activity converts urate to non-toxic allantoin—but humans do not express uricase. Human physiological levels of urate are clearly toxic to the tissues of rodents, as they promote oxidative stress via NADPH oxidase activation.91–93
Increased urate levels in humans, in addition to posing a risk for gout or gouty nephropathy, constitute a well-established risk factor for coronary disease, hypertension, type 2 diabetes and heart failure, as confirmed by meta-analyses94–98—although its impact often appears weak when other risk factors associated with metabolic syndrome are corrected for. Whether uric acid is a mediating risk factor in these disorders is very much in dispute. Speaking in favour of this view are studies demonstrating that xanthine oxidase inhibition with allopurinol often favourably influences endothelial dysfunction99–101; however, a counterargument is that xanthine oxidase activity generates superoxide, so allopurinol may simply be functioning as an antioxidant in these circumstances.102
Moreover, several studies in which urate levels have been modulated acutely by measures other than xanthine oxidase inhibition—raising it with an intravenous infusion, lowering it with an infusion of urate oxidase—have failed to observe any adverse impact of urate on endothelial function or other cardiovascular indices.103 ,104 Indeed, urate infusion was found to improve endothelial function in patients with type 1 diabetes, possibly reflecting the utility of urate as a peroxynitrite scavenger.105 The long-term marked elevation of urate with supplemental inosine—being studied in patients with multiple sclerosis as an antioxidant strategy—failed to influence blood pressure.106 Perhaps most compellingly, a number of recent Mendelian randomisation analyses, focusing on polymorphisms of renal tubular transport proteins for urate that influence serum urate levels, have failed to observe any impact of these polymorphisms on risk for heart disease, subclinical atherosclerosis, diabetes, hypertension, metabolic syndrome or diabetes107–111—with the exception of one small study targeting an Amish population which saw an association with blood pressure.112 The overall conclusion of these studies is that obesity and metabolic syndrome raise the serum urate level, and that the former, rather than urate per se, mediates the increased risk associated with elevated urate levels. The hyperinsulinaemia associated with metabolic syndrome promotes renal retention of urate, explaining at least in part the hyperuricaemia that is a feature of this syndrome.113 It appears that primates have evolved resistance to the pro-oxidant effects of urate demonstrated in rodents, such that losing their uricase activity did not compromise their Darwinian viability.
Hence, the failure of glycine to address the fructose-mediated elevation of serum urate levels, while unfortunate from the standpoint of gout risk, may not be disadvantageous from the standpoint of vascular health. Elevated urate levels appear likely to provide some protection from Parkinson's disease—a finding confirmed by a Mendelian randomisation analysis.114 ,115
References
Footnotes
-
Contributors MFM and JJD came up with the idea for the manuscript. MFM wrote the first draft. JJD reviewed, edited and contributed to concepts in the manuscript.
-
Competing interests None.
-
Provenance and peer review Not commissioned; internally peer reviewed.