Article Text

Abstract
Objective Circulating CD34+CD45− cell concentrations are increased in patients with coronary artery disease, however their pathophysiological significance is unknown. We determined CD34+CD45− cell concentrations following percutaneous coronary intervention (PCI) in order to explore their role in acute vascular injury.
Methods In a prospective time-course analysis, we quantified using flow cytometry circulating CD34+CD45− cells, traditional CD34+VEGFR-2+ putative endothelial progenitor cells (EPCs), CD14+ VEGFR− 2+Tie-2+ angiogenic monocytes and intercellular adhesion molecule expression (CXCR-4 and CD18) in patients, before and during the first week following diagnostic angiography (n=13) or PCI (n=23). Vascular endothelial growth factor-A (VEGF-A) and C reactive protein (CRP) were quantified by ELISA.
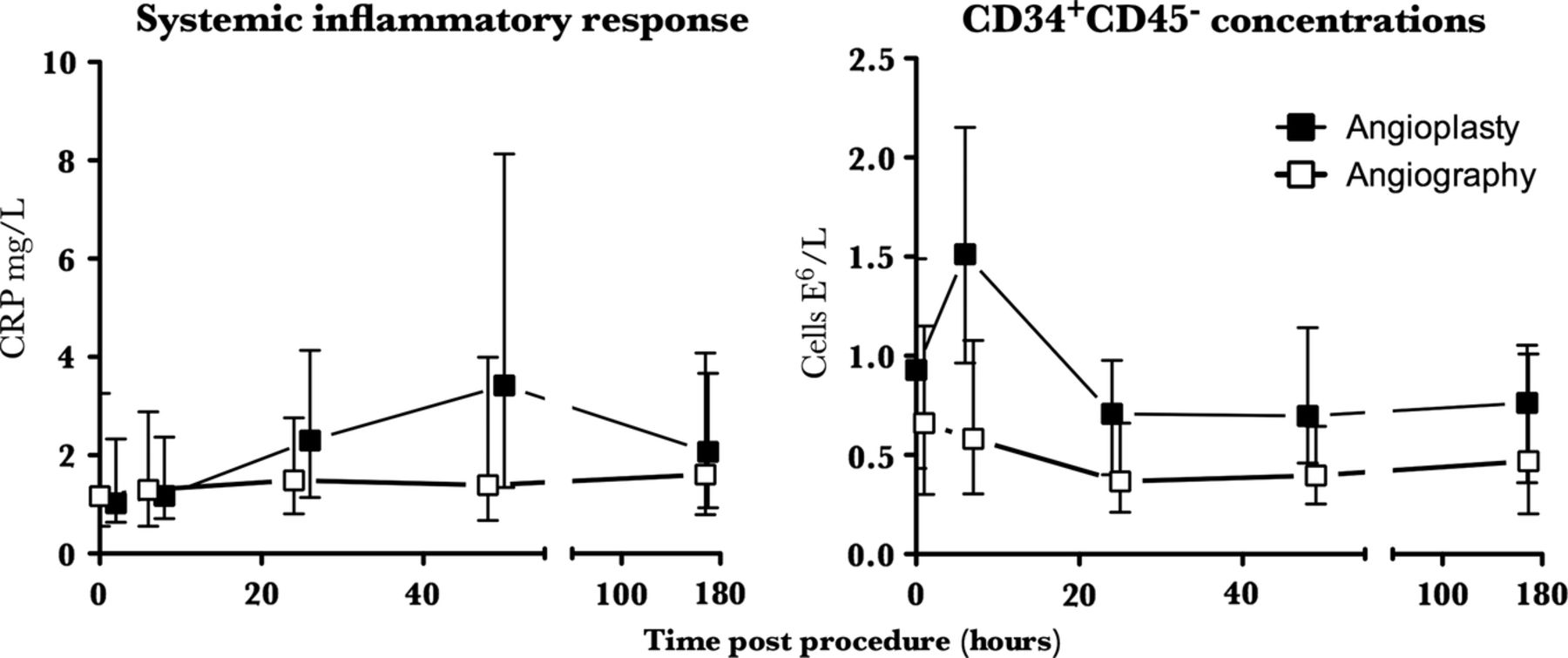
Results Unlike diagnostic angiography, PCI increased circulating neutrophil and CRP concentrations at 24 and 48 h, respectively (p<0.002 for both). CD34+CD45− cell concentrations were unaffected by angiography (p>0.4), but were transiently increased 6 h following PCI (median (IQR) 0.93 (0.43–1.49) vs 1.51 (0.96–2.15)×106 cells/L; p=0.01), returning to normal by 24 h.
This occurred in the absence of any change in serum VEFG-A concentration, adhesion molecule expression on CD34+ cells, or mobilisation of traditional EPCs or angiogenic monocytes (p>0.1 for all).
Conclusions PCI transiently increases circulating CD34+CD45− cells, without increasing CD34+ adhesion molecule expression or VEGF-A concentrations, suggesting that CD34+CD45− cells may be mobilised from the vessel wall directly as a result of mechanical injury. Traditional putative EPC and angiogenic monocytes are unaffected by PCI, and are unlikely to be important in the acute response to vascular injury.
- CORONARY ARTERY DISEASE
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 3.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/3.0/
Statistics from Altmetric.com
Key messages
What is already known about the subject?
-
Consistent with preclinical studies, we have previously reported an association between increased CD34+CD45– cell concentrations and coronary artery disease. The clinical significance and the source of this population of cells remain unclear. Conflicting data exist regarding the clinical relevance of traditional CD34+VEGFR-2+ endothelial progenitor cells (EPCs) and their role following acute vascular injury.
What does this study add?
-
Consistent with our previous findings, we observe in the present study that CD34+CD45– are rapidly mobilised following percutaneous coronary intervention (PCI). The functional significance and source of this CD34+CD45– population requires clarification, however it is likely that these cells are derived from the vessel wall. In support of previous work, traditional putative EPCs were unaffected by PCI and as such it appears unlikely they are important in the acute response to vascular injury.
How might this impact on clinical practice?
-
A better understanding of vascular progenitor cells may lead to therapies aimed at enhancing vascular repair in the context of atherosclerotic disease and following angioplasty.
Introduction
Percutaneous coronary intervention (PCI) strips the treated vessel of its endothelium and causes laceration of the underlying intima, initiating platelet aggregation and thrombus formation.1 This process triggers an inflammatory cascade that drives vascular smooth muscle proliferation and neo-intimal hyperplasia.2 The loss of endogenous thrombolytic function and anti-inflammatory signalling by the endothelium potentiates a maladaptive response to vascular injury that can lead to in-stent restenosis or thrombosis. Re-endothelialisation of the denuded arterial segment is therefore of crucial importance in protecting against adverse consequences following PCI. We have previously hypothesised that re-endothelialisation following PCI may be accelerated by circulating angiogenic monocytes and endothelial progenitor cells (EPC). EPCs are naive precursor cells that have the capacity to differentiate into mature endothelial cells and contribute to vascular regeneration.3
The traditional phenotypic definition of an EPC is based on the co-expression of VEGFR-2, and the stem cell marker CD34,4 ,5 either alone or in combination with CD133, a haematopoietic cell surface marker expressed only on very naïve cells.6 Circulating EPC concentrations are regulated by angiogenic growth factors such as vascular endothelial growth factor and stromal derived factor-1 acting through their cognate receptors, VEGFR-2 and CXCR-4.7–9 Peripheral homing and incorporation of EPCs are dependent on the expression of surface adhesion molecules, including β-integrins such as CD18.10 EPCs are mobilised in response to cardiovascular injury such as myocardial infarction,11 improve organ perfusion and function in animal models of ischaemia,12 and are negatively associated with adverse cardiovascular outcomes.13 ,14 Given their putative vasculoprotective role, it is plausible that EPC might be mobilised in order to facilitate vascular repair following injury. However, several investigators have shown previously that acute mobilisation of traditional phenotypic CD34+VEGFR-2+ EPCs in response to a discrete vascular injury during PCI does not occur.15 Mobilisation of CD34+VEGFR-2+ cells following PCI may however be delayed, as measurements have not been performed beyond 24 h. In a recent study involving a large cohort of over 200 patients undergoing coronary angiography, we observed that traditional EPC concentrations are unrelated to the severity of coronary atheroma, and are not increased following an acute coronary syndrome (ACS).16 This would indicate that traditional EPCs are not clinically relevant to atherosclerosis or acute atherothrombotic events. Indeed EPC, defined in the traditional fashion, are characterised by their capacity to form haematopoietic rather than endothelial colonies in culture, that lack significant proliferative potential or the capacity to incorporate into perfusing vessels in vivo.17 In contrast, non-haematopoietic CD34+CD45− cells form endothelial colonies with robust proliferative potential and can incorporate into perfusing vessels.17–19 Although not mobilised acutely by an ACS, we found that non-haematopoietic, CD34+CD45− concentrations, are directly associated with atheroma burden and also predict the occurrence of adverse coronary events.16 Circulating CD34+CD45− cells therefore serve as a measure of vascular injury, but their origin and pathophysiological significance are unknown. In particular, it is unclear whether or not they are bone marrow-derived cells with regenerative capacity.
Various alternate cell populations have been proposed as having vascular regenerative capacity. In particular, monocytes have received much attention. Monocytes accumulate at sites of new vessel formation and augment the differentiation and proliferation of endothelial cells via the secretion of pro-angiogenic factors.20 ,21 Vascular injury associated with PCI mobilises monocyte-derived angiogenic colony forming units (EC-CFU) within 24 h, indicating that this population participates in the acute cellular response to vascular injury.15 The specific subpopulations responsible for EC-CFU generation are unknown, however, monocytes expressing the endothelial epitopes vascular endothelial growth factor receptor-2 (VEGFR-2) and Tie-2, accelerate re-endothelialisation and improve left ventricular function following experimental cardiovascular injury,22–24 and are mobilised following an ACS.16 Their behaviour in response to discrete vascular injury, however, is unknown.
We conducted this study in order to determine the time course of circulating concentrations of a variety of putative angiogenic cell populations over a 1-week period following a discrete vascular injury caused by PCI.
Methods
Subjects
The study was performed with the approval of the local research ethics committee in accordance with the Declaration of Helsinki, and the written informed consent of all volunteers. Patients undergoing elective coronary angiography at Edinburgh Royal Infirmary, Edinburgh, UK, for the investigation of suspected angina (Canadian Cardiovascular Society grade 2 or more) or prior to valve replacement surgery were identified prospectively and invited to take part in the study. Patients with a recent ACS (<3 months), significant comorbid illness, haematological or internal malignancy, hepatic or renal failure or concurrent infection were excluded from the study. Clinical characteristics and medical therapy during admission were documented.
Coronary angiography
Coronary angiography was performed via the femoral or radial artery using 5–6F arterial catheters and standard angiographic projections. Stenoses affecting a major epicardial artery of ≥50% were defined as significant, and the overall atheroma burden was graded using the Gensini scoring system.25 Patients underwent PCI at the discretion of the operator following pretreatment with oral clopidogrel 300 mg and 5000 IU of intravenous unfractionated heparin. Intracoronary stents were implanted following lesion optimisation by balloon predilation and intracoronary glyceryl trinitrate.
Blood sampling and assays
Prior to coronary angiography, peripheral venous blood anticoagulated with ethylene diamine tetra-acetic acid (Sarstedt-Monovette, Germany) was collected for flow cytometry in all subjects. Whole blood was analysed for the differential leucocyte count using an autoanalyser (Sysmex, UK). Plasma troponin concentrations were measured using the ARCHITECT STAT troponin I assay (Abbott Laboratories, Abbott Park, Illinois, USA). An ELISA was used to quantify serum vascular endothelial growth factor A (VEGF-A) concentration according to the manufacturer's instructions (Invitrogen).26 Serum high-sensitivity C reactive protein (CRP) was quantified using an immunoturbidimetric method (Dade-Behring Marburg, Germany).27
Flow cytometric identification of EPCs
EPCs were phenotyped using flow cytometry as previously described.26 In brief, cells were stained directly and analysed for phenotypic expression of surface proteins using preconjugated antihuman monoclonal antibodies; anti-CD45-PercP (Becton Dickinson, Oxford, UK); anti-CD34-FITC; anti-VEGFR-2-APC; anti-VEGFR-2-PE; anti-Tie2-APC; anti-CXCR-4-APC; anti-CD18-PE (R&D systems, Minneapolis, USA); anti-CD-133-PE (Miltenyi Biotec Ltd, Surrey, UK); and anti-CD14-FITC (Caltag Systems, Buckingham, UK). Flow-cytometric analysis identified leucocytes by their characteristic forward and side scatter profile (figure 1). For each sample, approximately 500 000 events were acquired in the leucocyte gate using a FACS-Calibur flow cytometer (Becton Dickinson, UK). Data were analysed using FlowJo (Treestar). For quantification of subpopulations, the proportion of leucocytes bearing each epitope was determined individually using side scatter profile against the appropriate fluorescence channel, and permutations of co-expression were determined automatically using Boolean principles. Unstained samples served as negative controls (figure 1). Absolute cell concentrations per millilitre of blood were calculated by equating the total number of events in the leucocyte gate to the total leucocyte count.

Flow cytometric analysis of putative progenitor cells. Representative dot plots of negative controls and stained samples are shown in red and blue respectively. First, leucocytes were identified on the basis of their characteristic forward and side scatter profile (A). CD34-FITC (B and C) expression and the proportion of CD34+ events expressing CD18 (D and E) and CXCR-4 (F and G) were determined. Similarly CD133-PE+ (H and I), CD45-PercP+ (J and K), and VEGFR-2-APC+ (L and M) events were identified. Co-expression of surface markers was determined using Boolean principles. In separate analyses CD14-FITC+ events were identified (N), and those expressing Tie-2-APC and or VEGFR-2-PE were determined using quadrant analysis (O and P). Gates were set on single or unstained negative controls where appropriate.
Data analysis and statistics
Continuous variables are reported as the mean±SD or median and inter-quartile range where appropriate. Statistical analyses were performed with SPSS V.17 (SPSS Inc, Chicago, USA). The Shapiro-Wilk test was used to test for normality of distribution. Analyses were performed using parametric or non-parametric analysis of variance (ANOVA), where appropriate. The two-tailed Student t test, Mann-Whitney tests and Pearson's χ2 tests for between and within group comparisons were performed where appropriate. Statistical significance was taken at a two-sided p value of 0.05. At a significance level of 5% and based on power calculations derived from previous studies, a sample size of n=20 will give 80% power of detecting a twofold increase in traditional phenotypic EPC (CD34+VEGFR-2+ cells).
Results
Subjects and procedures
Thirteen patients underwent diagnostic coronary angiography alone and 23 underwent PCI using intracoronary stents. Patients were well matched in terms of age and sex and with respect to their cardiovascular risk profile and baseline medications (table 1). A diagnosis of hypertension was more frequently present in patients undergoing PCI compared to diagnostic coronary angiography alone (46% vs 83%; p=0.02). Patients undergoing diagnostic coronary angiography alone were more likely to have left main stem or three-vessel disease (p=0.001), although Gensini scores were similar (26 (16–80) vs 34 (26–79) units; p>0.70). Patients undergoing PCI required a longer total procedure time compared to diagnostic angiography alone (36±2.2 vs 16±1.6 min; p<0.001). Patients received a median of 1 (1–2) stent with a median total balloon inflation time of 44 (30–54) seconds. Drug eluting stents were used in 57% of patients (table 2). No significant complications arose during the course of the study and all patients were discharged within 24 h of the procedure.
Demographic, clinical characteristics and medical therapy of the study population
Procedural characteristics of the study population
Measures of inflammation and myocyte necrosis
All parameters were similar in both groups at baseline (p>0.05 for all; table 3). The median troponin concentration at 24 h was undetectable in both groups (p=0.13). Following PCI, the leucocyte count was increased (p=0.04 for ANOVA of repeated measures), peaking at 24 h (7.21±0.28 vs 6.24±0.20×106 cells/L; p=0.002). Serum CRP also increased following PCI (p=0.002 for ANOVA of repeated measures), peaking at 48 h (3.95 (1.26–8.26) vs 1.02 (0.64–2.33) mg/L; p=0.001). Serum VEGF-A concentrations were unchanged following PCI (p=0.16 for ANOVA of repeated measures). Diagnostic coronary angiography alone had no effect on the total leucocyte count, CRP or serum VEGF-A concentration throughout the study period (p>0.5 for ANOVA of repeated measures for all).
Inflammatory response following diagnostic angiography and percutaneous coronary angiography
Progenitor cell populations
All cell concentrations were similar in both groups at baseline (p>0.2 for all; table 4). Following PCI circulating CD34+CD45− cell concentrations were increased (p=0.0001, for one-way ANOVA of repeated measures), peaking at 6 h before returning to baseline concentrations at 24 h (1.51 (0.96–2.15) v 0.93 (0.43–1.49)×106 cells/L; p=0.01; table 4 and figure 2). The CD34+CD45− concentration was unaffected by diagnostic coronary angiography alone (p=0.43 for ANOVA of repeated measures; table 4 and figure 2). Traditional EPC phenotypes expressing CD34+VEGFR-2+ and those expressing CD34+VEGFR-2+CD133+ and CD34+CD45+ were unaffected in both groups (p>0.2 for all; table 4). CXCR-4 and CD18 expression on CD34+ cells was 47±7% and 36±5%, respectively, at baseline and were unchanged throughout the study period in both groups (p>0.2 for all; data not shown). The total CD14 count was not affected by either procedure (p>0.5 for both). Angiogenic monocytes expressing Tie-2 and VEGFR-2 comprised 0.72% (0.28–2.65) of circulating CD14+ cells and circulating concentrations were unchanged throughout the study in both groups (p>0.2 for all; table 4).
Cell frequencies following diagnostic angiography and percutaneous coronary intervention
{kind=link}
{kind=link}
Inflammatory response and CD34+CD45− cell release following percutaneous coronary intervention (PCI). PCI rapidly but transiently mobilised CD34+CD45− cells into the peripheral circulation, peaking at 6 h; p=0.01. Circulating concentrations had fallen back to baseline by 24 h. In contrast, the systemic inflammatory response caused by PCI was relatively prolonged, with CRP concentrations continuing to increase for at least 48 h; p=0.001. Diagnostic coronary angiography alone did not cause an inflammatory response or effect the CD34+CD45− concentration; p>0.5 for all. Data are median and IQR.
Discussion
We have examined the behaviour of a variety of circulating angiogenic cells in response to a discrete vascular injury caused by PCI. We observed a rapid but transient mobilisation of CD34+CD45− cells into the peripheral circulation following PCI, with circulating concentrations returning to baseline by 24 h. The systemic inflammatory response caused by PCI however, was relatively prolonged and continued to increase for at least 48 h. CD34+CD45− cells were mobilised without a detectable change in CD34+ surface adhesion molecules or serum VEGF-A concentration. Taken together, these observations would suggest that mobilisation of CD34+CD45− cells following PCI is not mediated by a circulating cytokine or growth factor acting on the bone marrow. Given the close association between CD34+CD45− and atheroma burden that we have previously observed,16 it is plausible that mechanical injury causes CD34+CD45− cells to be released from the coronary artery directly. Traditional phenotypic EPC populations (CD34+VEGFR-2+ and CD34+VEGFR-2+CD133+), and haematopoietic CD45+ subpopulations including angiogenic monocytes were both unaffected by discrete vascular injury up to 1 week following PCI. This would suggest that these populations are not mobilised in acute response to vascular injury and are unlikely to play a direct role in vascular repair; however, it remains possible that local cytokine production might increase adhesion of circulating proangiogenic monocytes or CD34+ cells to increase incorporation of these cell types to sites of vascular injury. Two small clinical studies have demonstrated a fall in CD34+VEGFR-2+ cells within the first few hours of coronary angioplasty. Neither of these studies used a control group, and interpretation is therefore limited, but it is likely that this ‘dip’ in CD34+ cell concentration is explained by the diurnal variation that is recognised to affect these populations,26 rather than incorporation to sites of vascular denudation. Previous studies have not specifically examined the behaviour of CD34+CD45− cells following PCI. Pelliccia et al28 found that higher concentrations of circulating CD34+VEGFR-2+CD45− and CD133+VEGFR-2+CD45− cells prior to revascularisation identified patients more likely to develop restenosis. Similarly, a direct relationship exists between coronary atheroma burden and both CD34+CD45− cells16 and CD34+CD45− cell-derived late outgrowth colonies.29 CD34+CD45− cells are therefore a measure of vascular injury; however, whether or not they are derived from the bone marrow or possess reparatory function remains unclear. Mature endothelial cells also express CD34+30 and are CD45−, and circulating CD34+CD45− cells may therefore simply reflect shedding of vascular detritus to the peripheral circulation as a consequence of cell turnover in atherosclerotic arteries. The early and transient appearance of CD34+CD45− cells in the circulation following vascular perturbation observed in the current study is consistent with this hypothesis. Furthermore, despite mobilisation of CD34+CD45− cells, neither plasma VEGF concentration nor the expression of the surface receptors, CXCR-4 or CD18 on circulating CD34+ cells were affected by PCI. It is possible that we have observed progenitor cell mobilisation from either the marrow or a marginated, vascular resident population and have failed to detect the circulating factors responsible for their release. Stem cell mobilisation occurs via activation of the phosphatidylinositol 3-kinase/Akt/endothelial nitric oxide synthase (PI3K/Akt/eNOS) pathway via angiogenic factors such as VEGF, through the stimulation of nitric oxide synthesis by bone marrow stromal cells.8 Increased nitric oxide bioavailability leads to cleavage of intracellular adhesions between stem cells and stromal cells of the bone marrow,31 allowing them to mobilise to the peripheral circulation in response to a stromal cell-derived factor-1 gradient generated by bone marrow stromal cells acting through the CXCR-4 receptor.7 ,9 Cell surface adhesion molecules such as CD18 mediate EPC homing and cells adhesion and are required for incorporation of EPCs into the vasculature.10 Although we have not performed a comprehensive evaluation of the factors involved in the mobilisation of stem cells from the bone marrow, the absence of any change in VEGF concentration or CD18 and CXCR-4 expression further supports the hypothesis that CD34+CD45− cells were not mobilised from the bone marrow, but released directly from the injured coronary artery.16 ,32
While we did not observe an increase in CD34+CD45− cells in the present study, this has been described following PCI, previously. However, these studies did not evaluate vascular injury in isolation and included patients with significant myocardial necrosis following acute myocardial infarction.33 ,34 Peak mobilisation varied from 24 h to a week, and was proportional to the degree of systemic inflammation with CD34+CD45− concentrations predicting future development of in-stent restenosis. Whether CD34+CD45− cells contribute directly to the development of restenosis is unknown, although it is interesting to note that the use of drug-eluting stents was associated with less inflammation and CD34+CD45− mobilisation. This suggests drug-eluting stents may in part suppress restenosis by attenuating inflammatory typo (signaling) to the bone marrow by suppressing local vascular inflammation. CD34+CD45− cells have the capacity to adopt smooth muscle cell characteristics in vitro and may therefore potentiate the development of in-stent restenosis by acting as circulating smooth muscle progenitor cells.35 Putative smooth muscle progenitor cells have been reported to express both CD34 and VEGFR-2,36 and both CD34 and CD133 are expressed at higher concentrations in the neo-intima of restenotic lesions compared with de novo lesions.37 Inoue et al33 demonstrated that circulating mononuclear cells of patients with in-stent restenosis exhibit a propensity to develop a smooth muscle phenotype over that of an endothelial phenotype compared with those without. Therefore, plasticity of circulating CD34+ cells may lead to acceleration of neo-intimal hyperplasia if a pro-inflammatory microenvironment in a particular patient dictates such a response.
Monocytes will express a variety of endothelial characteristics in-vitro under appropriate angiogenic stimulation.20 ,38 ,39 Monocyte chemoattractant protein-1 (MCP-1) is released by endothelial cells in response to shear stress and tissue ischaemia,40 causing monocytes to accumulate at sites of new vessel formation, adhere to injured endothelium, accelerate re-endothelialisation and improve endothelial vasomotor function.22 ,23 However, the remarkable plasticity of monocytes may result in a diverse response to vascular injury depending on the local microenvironment. For instance, there is a strong correlation between circulating monocyte concentration and the development of in-stent restenosis,41 and MCP-1 is significantly elevated in both plasma42 and coronary atherectomy specimens of patients who develop in-stent restenosis following PCI.43 Furthermore, treatment with anti-MCP-1 monoclonal antibodies following balloon angioplasty in rats inhibits neo-intimal hyperplasia.44 The role of monocytes following vascular injury is therefore diverse and again probably depends on the local microenvironment. PCI mobilises EC-CFU within 24 h although whether specific subpopulations participate in the acute cellular response to vascular injury is unknown.15 We recently observed that monocytes expressing Tie-2 and VEGFR-2 are mobilised acutely following an ACS; however, this appeared to be a concequence of myocyte necrosis rather than the presence of atheroma burden or the presence of an ACS, per se.16 In the present study, CD14+Tie-2+VEGFR-2+ cells were not mobilised following PCI, suggesting that mobilisation of this monocytic subpopulation occurs in response to more extensive myocardial injury rather than discrete vascular injury.
Study limitations
Although we hypothesise that CD34+CD45− cells have been liberated directly from the coronary artery, we have not proven this definitively and further confirmatory studies are required. Patients undergoing PCI were more frequently hypertensive. Hypertension has been linked to impaired mobilisation of ‘EPC’ and related cell populations, and this may therefore be relevant to our results.45 If PCI-mediated vascular injury does mobilise traditional EPC, this effect might be muted in a group of hypertensive patients, potentially leading to a type 2 error. However, previous investigators have demonstrated the absence of mobilisation of traditional EPC in a group of largely non-hypertensive patients.46 Patients undergoing PCI were all administered heparin, which has been shown to increase circulating CD34+ cell concentrations, possibly through disruption of SDF/CXCR-4 interaction.47 Heparin may have led to CD34+CD45− mobilisation; however, if this were the case one would expect the CD45+ fraction to be similarly increased.
Conclusions
Discrete vascular injury associated with PCI causes a rapid but transient release of CD34+CD45− cells into the peripheral circulation in the absence of a concurrent increase in CD34+ adhesion molecule expression or VEGF-A secretion. Confirmatory studies are required to determine whether CD34+CD45− cells released into the circulation following PCI arise directly from the vessel wall through mechanical injury, or are progenitors with reparatory capacity mobilised from a stem cell niche. Traditional EPC and VEGFR-2+Tie-2+ monocytes are unaffected by PCI, and are unlikely to be important in the acute response to vascular injury.
References
Footnotes
-
Contributors All authors contributed significantly to the work. GJP and NLM devised and conducted the research and wrote the manuscript along with DEN. EF assisted with data acquisition and analyses. OT-C, GRB and MT assisted with data analysis and manuscript preparation. GJP and NLM take overall responsibility for the manuscript.
-
Funding The research was supported by a BHF Project Grant (PG/07/017/22405). The Wellcome Trust Clinical Research Facility is supported by NHS Research Scotland (NRS) through NHS Lothian.
-
Competing interests A British Heart Foundation (BHF) Scholarship (SS/CH/92010) and Intermediate Clinical Research Fellowship (FS/10/024) supported GJP and NLM, respectively. DEN is supported by the British Heart Foundation (CH/09/002).
-
Ethics approval Lothian Research Ethics Committee.
-
Provenance and peer review Not commissioned; externally peer reviewed.
-
Data sharing statement Technical appendix, statistical code and dataset are available on request. Explicit informed consent for data sharing was not given but the risk of identification is low.