Article Text

Abstract
Background The cardiometabolic effects of SRT2104, a novel SIRT1 activator, were investigated in people with type 2 diabetes mellitus (T2DM).
Methods Fifteen adults with T2DM underwent a randomised, double-blind, placebo-controlled cross-over trial and received 28 days of oral SRT2104 (2.0 g/day) or placebo. Forearm vasodilatation (measured during intrabrachial bradykinin, acetylcholine and sodium nitroprusside infusions) as well as markers of glycaemic control, lipid profile, plasma fibrinolytic factors, and markers of platelet-monocyte activation, were measured at baseline and at the end of each treatment period.
Results Lipid profile and platelet-monocyte activation were similar in both treatment arms (p>0.05 for all). Forearm vasodilatation was similar on exposure to acetylcholine and sodium nitroprusside (p>0.05, respectively). Bradykinin-induced vasodilatation was less during treatment with SRT2104 versus placebo (7.753vs9.044, respectively, mean difference=−1.291,(95% CI −2.296 to −0.285, p=0.012)). Estimated net plasminogen activator inhibitor type 1 antigen release was reduced in the SRT2104 arm versus placebo (mean difference=−38.89 ng/100 mL tissue/min, (95% CI −75.47, to –2.305, p=0.038)). There were no differences in other plasma fibrinolytic factors (p>0.05 for all).
After 28 days, SRT2104 exposure was associated with weight reduction (−0.93 kg (95% CI −1.72 to −0.15), p=0.0236), and a rise in glycated haemoglobin (5 mmol/mol or 0.48% (0.26 to 0.70), p=0.004)
Conclusions In people with T2DM, SRT2104 had inconsistent, predominantly neutral effects on endothelial and fibrinolytic function, and no discernible effect on lipids or platelet function. In contrast, weight loss was induced along with deterioration in glycaemic control, suggestive of potentially important metabolic effects.
Clinical trial registration NCT01031108; Results.
- sirtuins
- SIRT-1 activator
- type 2 diabetes mellitus
- weight loss
- lipids
- endothelial dysfunction
- lipids
- cardiovascular risk
- platelet activation
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
- sirtuins
- SIRT-1 activator
- type 2 diabetes mellitus
- weight loss
- lipids
- endothelial dysfunction
- lipids
- cardiovascular risk
- platelet activation
Key Questions
What is already known about this subject?
Sirtuins are a group of proteins that regulate many important biological pathways. Specifically, SIRT1 mediates the beneficial effects of caloric restriction on endothelial function and on glucose regulation in animal models. The cardiometabolic effects of SIRT1 activation in humans are unknown; it is this research question that underpins this randomised clinical trial.
What does this study add?
It appears that short-term SIRT1 activation in humans is well tolerated, and has predominantly neutral effects on markers of endothelial function and platelet-monocyte function. However, it is associated with weight reduction and a rise in glycated haemoglobin.
How might this impact on clinical practice?
The reduction in weight suggests that SIRT1 activation indeed modulates important biological pathways related to energy metabolism. People with type 2 diabetes mellitus have a high prevalence of obesity, and this finding of weight loss merits closer investigation. The rise in glycated haemoglobin is not consistent with results from animal model studies. We speculate that the deterioration in glucose control may be a signal of inadequate reduction in insulin resistance in humans. Further studies are required to confirm this signal, which will impact on the feasibility of development of this novel class of drugs.
Introduction
The effect of caloric restriction on energy metabolism has attracted intense interest over the past decade because it prolongs the life span in yeast, nematodes and flies. Caloric restriction also lowers the incidence of age-related disorders such as diabetes and cancer in mammals.1–5 These beneficial effects may be mediated by sirtuins, (‘SIR-2-ins’ or silent mating-type information regulation 2). In yeast, the sirtuin gene regulates many cellular pathways including inflammation, apoptosis and mitochondrial biogenesis. The activity of sirtuins is dependent on nicotinamide adenine dinucleotide , thereby linking energy metabolism and caloric restriction to these important cellular pathways.6
The mammalian homologue of the sirtuins is SIRT1.7 In murine models, activation of SIRT1 has been implicated in the modulation of several cellular substrates including peroxisome proliferator-activated γ cofactor 1-α, nuclear factor κ-B, foxhead box protein and uncoupling proteins.8–10 These pathways are implicated in skeletal muscle energy metabolism and adipocyte differentiation, and play a role in glucose regulation in skeletal muscle and pancreatic β cells, as well as modulating insulin sensitivity.11 Caloric restriction also improves endothelial function in rodent and human studies through weight loss, improvement of nitric oxide production and reduction in reactive-oxygen species.12–14 Inhibition of SIRT1 promotes atherogenesis and upregulation of SIRT1 increases nitric oxide in rodent models.15–17 As a caloric restriction mimetic, SIRT1 activation therefore has the potential to improve cardiovascular health and mitigate the adverse effects of atherosclerotic disease and its risk factors.
Type 2 diabetes mellitus (T2DM) is a highly prevalent chronic condition, estimated to affect 4.4% of the worldwide population by 2030.18 It is associated with an increasing economic burden and in the USA total costs are estimated to be $245 billion.19 The major cause of mortality in patients with T2DM is cardiovascular disease. One of the putative mechanisms of the link between cardiovascular disorders and T2DM is endothelial dysfunction.20 Thus, modulation of SIRT1 is a promising strategy for the treatment of both diabetes and its well documented association with cardiovascular disease.
To date, studies have primarily investigated the ex vivo effect of SIRT1 activation. We hypothesised that therapeutic SIRT1 activation would have beneficial effects on cardiometabolic health in people with diabetes. Specifically, we examined the effect of a novel SIRT1 activator, SRT2104, on vascular vasomotor and fibrinolytic function, platelet activation, lipid profile and markers of glycaemic control in people with T2DM.
Methods
The study was approved by the research ethics committee, was given clinical trial authorisation by the Medicines and Healthcare products Regulatory Agency (MHRA), and carried out at the MHRA phase I accredited Wellcome Trust Clinical Research Facility at the Western General Hospital, UK. Written informed consent was obtained from each participant, and the study was carried out in accordance with the Declaration of Helsinki.
Study participants
Fifteen individuals with T2DM were recruited from outpatient clinics at the Royal Infirmary of Edinburgh. Inclusion criteria included age between 18 years and 70 years, glycated haemoglobin (HbA1c) <9.0% (75 mmol/mol) and resting blood pressure of <160/90 mm Hg. Exclusion criteria included current smokers, the use of ACE inhibition therapy (as the potentiation of bradykinin and effects on endothelial function would confound the vascular studies), the presence of major comorbidities, chronic illness, renal or liver impairment, history of gastrointestinal diseases or surgeries influencing drug absorption, history of alcoholism, history of neoplastic disease within the last 5 years, a positive urinary test for recreational drugs, pregnancy, and participation in other clinical trials or blood donation within the last 3 months. Eligibility of participants including absence of relevant medical history was confirmed through a standardised form completed by the subject’s primary care physician after obtaining informed consent. Tests for pregnancy (serum human chorionic gonadotropin (HCG) concentrations at screening and urinary HCG concentrations at study visits) were conducted on all female participants of childbearing potential.
Study design
This was a prospective, double-blind, randomised, placebo-controlled cross-over study. Subjects were randomised 1:1 to receive 2.0 g daily of oral SRT2104 or matched placebo (Sirtris Pharmaceuticals) for a 28-day period, followed by cross-over to the alternate study arm for another 28 days, giving a total dosing duration of 56 days. A safety visit was conducted on day 70, with a follow-up by telephone on day 86. Assessment of drug safety, tolerability and efficacy on vascular function was carried out at baseline, and during and at the end of each treatment period (figure 1). The overall study design included otherwise healthy smokers in addition to people with T2DM, and volunteers were stratified by these two categories. This manuscript focuses on the T2DM group, with observations in the otherwise healthy smokers group described previously.21
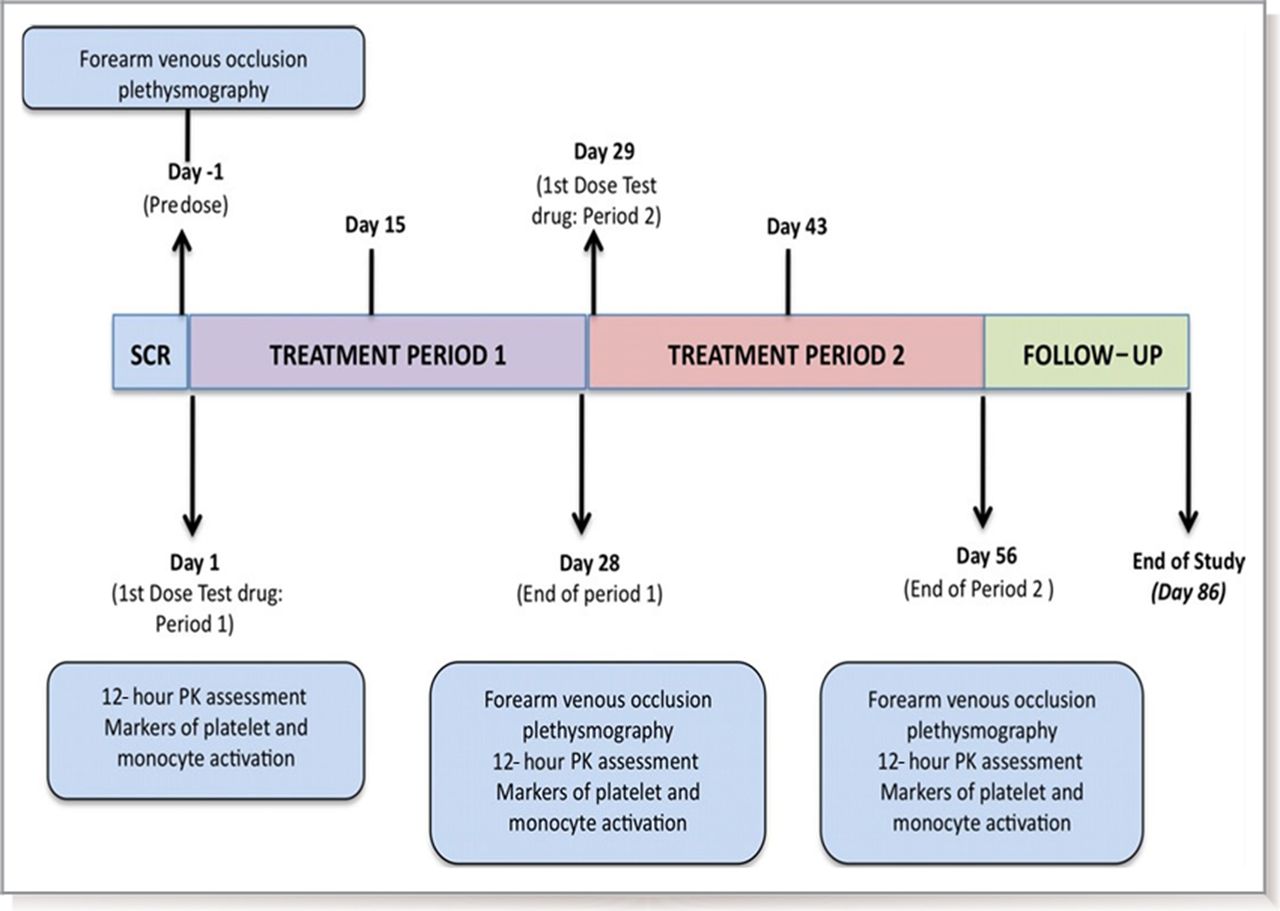
Study design: schematic representation of study design. PK, pharmacokinetic; SCR, screening.
Vascular studies
Vascular studies were undertaken before and at the end of each 28-day trial period. All studies were performed with the patient lying supine in a quiet temperature-controlled (22°C to 25°C) room. Participants were fasted for 10 hours, and avoided caffeine and alcohol for 24 hours, before the study. Venous cannulae (17G) were inserted into large subcutaneous veins in the antecubital fossae of both arms to facilitate periodic venous sampling. Supine heart rate and blood pressure were monitored at intervals throughout the study using a semiautomated, non-invasive oscillometric sphygmomanometer (Omron 705 IT).
Forearm venous occlusion plethysmography
Forearm blood flow was measured in the infused and non-infused forearms using forearm venous occlusion plethysmography as described previously.22 Volunteers underwent brachial artery cannulation in the non-dominant forearm with a 27 standard wire gauge steel needle. After a 20 min baseline infusion with 0.9% saline, incremental intra-arterial doses of bradykinin (American Peptide) at 100 pmol/min, 300 pmol/min and 1000 pmol/min (an endothelium-dependent vasodilator that induces tissue plasminogen activator (t-PA) release), acetylcholine (Chem. Pharm Fabrik) at 5 µg/min, 10 µg/min and 20 µg/min (an endothelium-dependent vasodilator that does not induce t-PA release) and sodium nitroprusside (Hospira) at 2 µg/min, 4 µg/min and 8 µg/min (an endothelium-independent vasodilator that does not induce t-PA release) were infused for 6 min at each dose, with a 30 min 0.9% saline washout infusion between drugs. The order of drugs was randomised between subjects but kept constant for each participant across the three visits.
Blood sampling
Paired venous blood samples were obtained from each forearm before and during the infusion of intra-arterial bradykinin. Samples were collected into acidified buffered citrate (Stabilyte; Trinity Biotech) and citrate (BD Vacutainer; BD UK) for determination of t-PA and plasminogen activator inhibitor type 1 (PAI-1) concentrations, respectively. Samples were placed on ice before centrifuging at 2000 g for 30 min at 4°C. Platelet-free plasma was decanted and stored at −80°C before further analysis. Venous blood samples were collected into EDTA at the beginning and end of the vascular study to determine haematocrit.
Plasma t-PA antigen and activity (t-PA Combi Actibind t-PA ELISA kit; Technoclone, Vienna, Austria) and PAI-1 antigen and activity (Elitest PAI-1 Antigen and Zymutest PAI-1 Activity; Hyphen Biomed) concentrations were determined by ELISAs.
Platelet and monocyte activation
Flow-cytometric measurements of platelet–monocyte aggregation and platelet surface expression of P-selectin and monocyte CD11b expression (Mac-1/CD11b) were performed at baseline and at the end of each treatment period as described previously.23–26 Briefly, peripheral venous blood was drawn from a large antecubital vein and anticoagulated with the direct thrombin inhibitor D-phenylalanine-L-arginine chloromethyl ketone (Cambridge Biosciences) and immunolabelled within 5 min of phlebotomy for subsequent flow cytometric analysis. Directly conjugated monoclonal antibodies were obtained from DakoCytomation and Serotec. Samples were stained with the following conjugated monoclonal antibodies: phycoerythrin (PE)–conjugated CD14, PE-conjugated CD62p, PE-conjugated CD11b, fluorescein isothiocyanate (FITC)–conjugated 42a and FITC-conjugated CD14 and appropriate control isotypes. Once stained, samples were incubated for 20 min at room temperature before being fixed with Fluorescence-activated cell sorting (FACS) Lyse (Becton-Dickinson). All samples were analysed using a FACS Calibur flow cytometer using CellQuest Pro software (Becton-Dickinson). Venous blood was collected in citrate at baseline and after each dosing period to assess plasma-soluble CD40 ligand concentrations. Blood was centrifuged at 1500 g for 15 min at 4°C, and plasma was decanted and stored at −80°C for further analysis by ELISA (Bender Medsystems).
Safety and pharmacokinetic analyses
Venous blood samples were collected biweekly to measure haematological and biochemical parameters including full blood count, coagulation profile, liver and renal function, creatine phosphokinase, lactate dehydrogenase, lipid profile and free fatty acids. Analyses were conducted by the regional clinical haematology and biochemistry reference laboratories using an automated haematology analyser (XE2100, Sysmex Corporation and ACL TOP, Instrumentation Laboratory), an automated chemistry analyser using colorimetric, kinetic and enzymatic ultraviolet and colour assays (AU2700/AU640 analysers, Beckman Coulter), ion-selective electrodes (sodium, potassium and chloride assays) and two-point and multiple-point rate assays (Ortho Clinical Vitros 250 analyser). Venous blood samples were taken into prelabelled heparinised sodium tubes for pharmacokinetic assessment of plasma SRT2104 concentrations (Simbec Laboratories Limited). Serial blood samples were collected on days 1, 28 and 56 immediately before (0 min) and 15 min, 30 min, 60 min, 120 min, 180 min, 240 min, 480 min, 720 min and 1440 min following study medication. Plasma was separated by centrifugation of whole blood at 1500 g at 4°C for 15 min, and decanted and stored at −80°C until analysed.
Plasma concentrations of SRT2104 were measured using liquid chromatography with tandem mass spectrometry detection in positive ion mode. High-pressure liquid chromatography was performed using Betasil silica–100 columns using a Phenomenex C18 guard column.27
Data analysis and statistics
Plethysmographic data were analysed as described previously.22 Estimated net release of t-PA and PAI-1 antigen and activity was defined as the product of forearm plasma flow (based on blood flow and haematocrit) and the difference in plasma antigen (or activity) concentrations between the two forearms. Fibrinolysis and forearm blood flow data were analysed using a linear mixed-model repeated-measures analysis of covariance. Treatment differences were investigated in a model adjusting for period, treatment by period, vasodilator dose, treatment by vasodilator dose, and vasodilator dose by period using SAS for UNIX (V.9.1.3 or higher; SAS Institute). Treatment differences in weight, HbA1c, fructosamine and lipid variables were analysed using linear mixed-model repeated-measures analyses of covariance adjusting for baseline, period and treatment by period interaction. Values for these parameters are expressed as model adjusted (least square) means and 95% CIs. Between-day reproducibility of forearm venous occlusion plethysmography data were assessed using the Bland-Altman method, and coefficient of reproducibility was determined for 95% CIs using the Student's t-distribution. All other values are expressed as mean±SD Statistical significance for treatment differences was concluded if the two-sided p was <0.05. Interactions and period effects were tested using a significance level of 0.10.
Results
The Consolidated Standards of Reporting Trials(CONSORT) flow diagram (figure 2) summarises the participant screening, exclusion and recruitment process. The participants were predominantly middle-aged men (58±7.8 years, 13 male), who were obese (body mass index 30±3.5 kg/m2) and who had reasonable glycaemic control (mean HbA1c 54±8.0 mmol/mol (7.4%±0.80%)).

CONSORT flow diagram of randomisation.
Safety
All of the participants tolerated the study medication. Commonly reported side effects occurring in two or more volunteers included headache (33%), diarrhoea (27%), nausea (13%) and hypoglycaemia (13%) (table 1). Apart from one subject describing diarrhoea as ‘severe’, the remaining reported adverse events were mild to moderate in intensity. All side effects resolved without any sequelae. There were no meaningful differences in the frequency of treatment-emergent adverse events between the active treatment and placebo groups. One individual underwent part of the first period of the study but was withdrawn because an adverse event criterion was met with the concentration of alanine aminotransferase being recorded at five times the upper limit of normal. After unblinding, this patient was found to have been taking placebo at the time of the event.
Summary of treatment-emergent adverse events. Multiple adverse events for each subject are counted once within each unique category
Pharmacokinetics of SRT2014
After 28 days of active treatment, the mean maximum plasma concentration (Cmax) of SRT2104 was 517±355 ng/mL and 716±359 ng/mL in the first and second treatment periods, respectively. The mean time at which the maximum plasma concentration was observed (Tmax) on day 28 of dosing was 2.6±1.2 hours and 2.9±1.5 hours in the first and second treatment periods, respectively. The mean area under the curve was 5300±3473 h.ng/mL in the first treatment period and 7312±3708 h.ng/mL in the second treatment period. The pharmacokinetics in this group was consistent with levels seen in previous studies.27
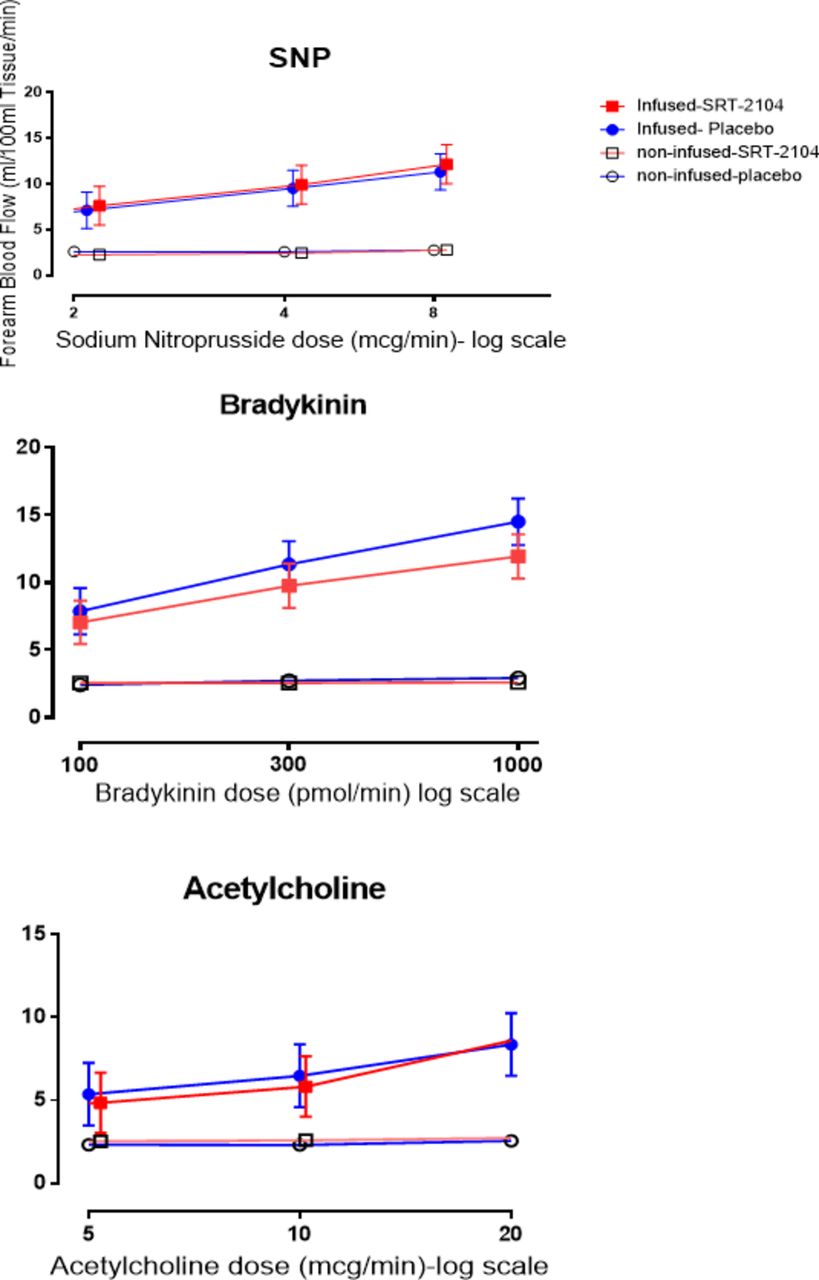
Cardiovascular effects of SRT2104
Blood pressure and heart rate remained unchanged throughout the study. No effects were observed on cardiac rhythm or the 12-lead ECG, and specifically the corrected and uncorrected QT intervals were unaffected. A dose-dependent increase in the infused forearm blood flow was observed with all three agonists (acetylcholine, bradykinin and sodium nitroprusside) in the presence of either SRT2104 or placebo (p<0.0001 for all three agonists; figure 3). There were no differences in response to acetylcholine (p=0.318) and sodium nitroprusside (p=0.083) in the presence of SRT2104 compared with placebo. There was a reduction in bradykinin-induced vasodilatation with SRT2104 (7.753 vs 9.044, SRT2104 vs placebo, mean difference=−1.291, (95% CI −2.296 to −0.285, p=0.012)) with a trend for a period-by-treatment effect (p=0.092).
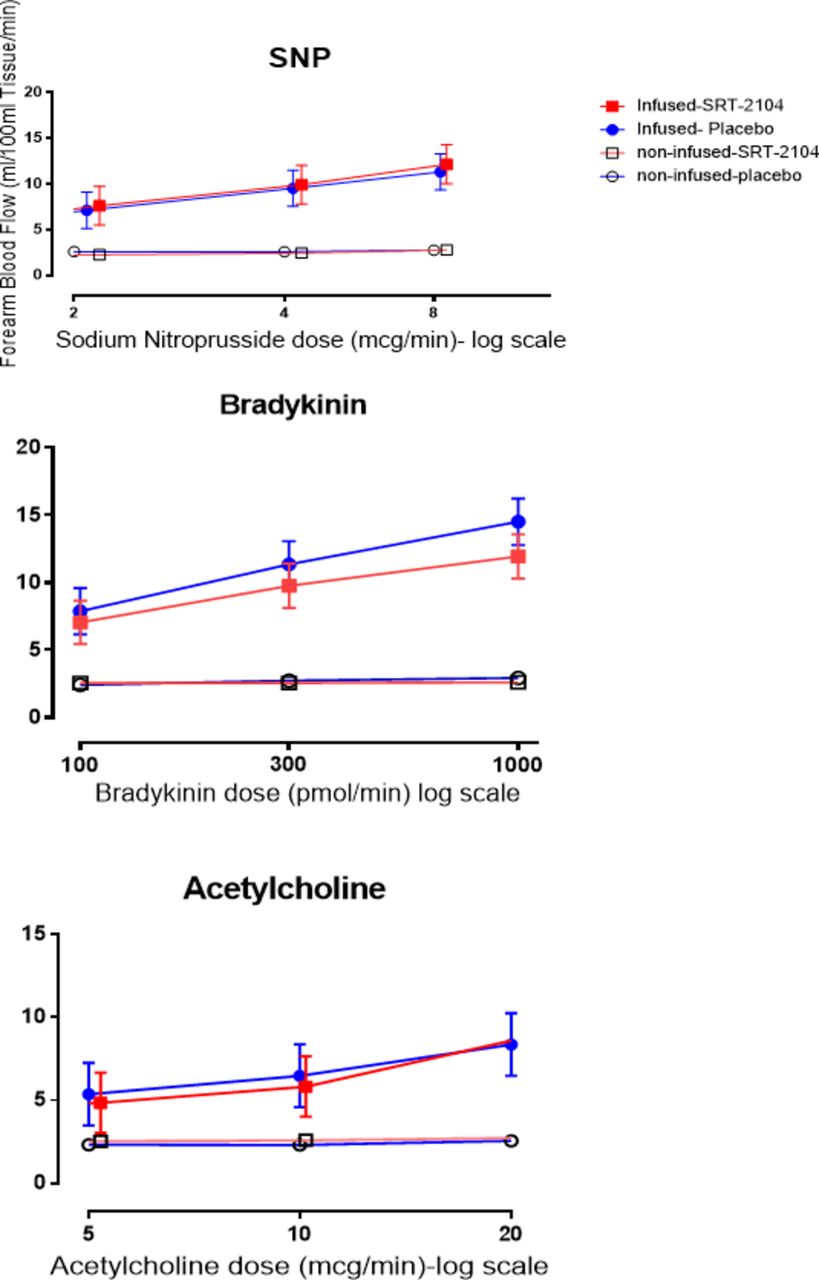
Effect of bradykinin (100 pmol/min, 300 pmol/min, 1000 pmol/min), acetylcholine (5 µg/min, 10 µg/min, 20 µg/min) and sodium nitroprusside (2 µg/min, 4 µg/min, 8 µg/min) on absolute forearm blood flow. Blue, placebo; red, SRT2104; closed circle and square, infused forearm blood flow; open circle and square, non-infused forearm blood flow. Data are presented as mean ±95% CI.
Endogenous fibrinolysis and monocyte and platelet activation
Post hoc analysis showed that dose-dependent increments were recorded in bradykinin-induced net t-PA antigen and activity release (p<0.0001 for both) in the infused arm. Estimated net PAI antigen release was reduced with SRT2104 compared with placebo (mean difference=−38.89 ng/100 mL tissue/min, (95% CI −75.47 to –2.305, p=0.038)) with a non-significant period effect for the plasma PAI-1 antigen concentrations (p=0.138). There were no differences in net PAI-1 activity release, or t-PA antigen and activity release (p>0.05 respectively). SRT2104 had no effect on markers of in vivo platelet or monocyte activation (figure 4).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Effect of SRT2104 on markers of platelet and monocyte activation. Data are presented as mean±SD. PMA, platelet-monocyte aggregate; CD11b, CD 11b/macrophage-1 antigen; sCD40L, soluble CD40 ligand.
Metabolic effects
During SRT2104 administration, body weight decreased by 0.93 kg (95% CI −1.72 to −0.15, p=0.0236) with a treatment-by-period effect (p=0.080). HbA1c rose by 0.48% (95% CI 0.26% to 0.70%, p=0.004) after 28 days of SRT2104, as did plasma fructosamine, which rose by 33.41 µmol/L (95% CI 20.24 to 46.58 µmol/L, p<0.001).
Post hoc analyses showed that the lipid profile did not change (table 2) although trends were noted towards lower concentrations of total cholesterol (−0.36 mmol/L, 95% CI −0.87 to 0.16 mmol/L, p=0.158) and triglycerides (−0.22 mmol/L, 95% CI −0.53 to 0.09 mmol/L, p=0.150; table 2) with SRT2104. Non-esterified fatty acids rose by 0.09 mmol/L (95% CI 0.04 to 0.15 mmol/L, p=0.003) with a treatment-by-period effect (p=0.0087).
Effect of SRT2104 on serum lipid concentrations
Discussion
The cardiometabolic effects of SRT2104 have not been studied previously in people with T2DM. Here we present the data in patients with T2DM that are broadly consistent with our previously reported observations in otherwise healthy cigarette smokers.21 However, we did note some important contrasts especially in metabolic parameters.
In otherwise healthy smokers, we have previously found no differences in forearm blood flow, platelet aggregation and fibrinolysis with SRT2104. Our findings in the current T2DM cohort were similar although we found modest reductions in bradykinin-induced vasodilatation and net PAI-1 antigen release with SRT2014. This finding is unexpected from previous animal models.15 17 We did observe some treatment-by-period interactions suggesting the potential for an inadequate washout period. Moreover, our sample size was modest and this could represent a chance finding. An alternative explanation is that SRT2104 affects a hitherto undescribed pathway which works via bradykinin alone and not acetylcholine or sodium nitroprusside. This study cannot answer that question presently but generates a hypothesis for further examination in the future. Once again in contrast to the smokers’ cohort we found a statistically significant decrease in PAI-antigen release. There was no significant treatment-by-period interaction in this case (p>0.1) which would suggest a potential fibrinolytic benefit in the treatment arm, although this was not confirmed by other measures of t-PA and PAI-1 activity, which were unaltered. Taken in aggregate, the effect of SRT2104 on cardiovascular measures are predominantly neutral although we acknowledge some inconsistent changes in isolated measures of vasomotor and fibrinolytic function.
Obesity is a major factor in the development of T2DM through the promotion of insulin resistance. Weight reduction is difficult to achieve in many patients and remains a major focus of therapeutic and lifestyle intervention. In the present study a striking reduction in weight over a 28-day period was observed that was not observed with matched placebo. It is unknown whether weight reduction with SRT2104 in overweight patients with T2DM would be sustained and have long-term benefits. Whether it could be attributable to appetite suppression through SIRT1 activation or a consequence of enhanced metabolic effects is unclear, but this is a potentially important observation that warrants further investigation. A treatment-by-period interaction was observed with this result as well, so this result should be interpreted with caution. If sustained and reproducible, this may represent a novel approach to the treatment of obesity.
The loss of weight was associated with an apparent short-term deterioration in measures of glycaemic control. The elevations of HbA1c and fructosamine were puzzling. These observations contradict the findings in models of mice and other higher mammals. It is possible that the acute administration of SRT2104 mimics the early changes of fasting states (decreased insulin secretion), and that the subsequent effects of decreased insulin sensitivity take longer to develop. This may explain the weight loss that was observed and to a certain extent the lack of benefit to glycaemic control. Supporting this theory, a trend towards decreased peak insulin secretion to a glucose challenge was observed in a separate study (2-hour postglucose challenge insulin concentrations 143 mmol/L vs 117 mmol/L, placebo vs SRT2104, p=0.046) where a similar degree of weight loss (approximately 1 kg in 28 days) was observed.28 It may be that in people with T2DM with established relative or absolute insulin deficiency, attenuation of insulin secretion, without a concomitant reduction in insulin resistance, is sufficient to permit a net rise in blood glucose.
Another potential putative mechanism linking SIRT1 modulation to an acute rise in insulin resistance is the inhibition of Peroxisome proliferator-activated receptor gamma (PPAR-γ) and the mobilisation of fat from white adipose tissue.29 This may be responsible for the modest rise in non-esterified fatty acids seen after SRT2104 administration that could contribute to the change in glycaemic control, leading to insulin resistance via intracellular competition with glucose metabolism.30
People with T2DM have complex changes to other important glucoregulatory hormones such as glucagon and cortisol.31 During acute starvation, important metabolic adaptations occur such as gluconeogenesis and an increase in glucocorticoids. These metabolic adaptations maintain the supply of blood glucose to the brain during periods of prolonged starvation, but result acutely in a constellation of effects similar to insulin resistance. However, in people with established T2DM, these protective glucoregulatory mechanisms described above may exacerbate hyperglycaemia in the short term.
Without further study, it is unclear whether long-term exposure to SRT2104 will ultimately be metabolically beneficial in patients with T2DM. The promotion of hyperglycaemia, which is undesirable, conflicts with the beneficial effects of weight loss, and it is unknown whether these effects are sustained over time. The finding of weight loss certainly raises the question as to whether the drug might be beneficial in individuals with impaired fasting glucose who are treatment-naive, and whether an exposure to the drug beyond 28 days would improve glycaemia once the downstream effects of increased mitochondrial activity and decreased adiposity are further established.
The principal aim of the present study was to establish whether SRT2014 could improve a range of markers of cardiovascular health in patients with T2DM. Ultimately, we did not demonstrate any improvements in vasomotor or fibrinolytic vascular function or measures of platelet and monocyte activation.
An important question was whether the dose of SRT2104 was sufficient to have an effect. As yet, the pharmacokinetics and pharmacodynamics of SRT2104 are not fully elucidated. However, an effect on metabolic measures was demonstrated with substantial exposure to SRT2104, achieving high plasma SRT2104 concentrations in the current study. Even if adequate SIRT1 activation is assumed, its downstream effects may vary in different tissues in different disease states. It may be that in advanced states of disease associated with ageing, such as T2DM, the beneficial effects are abolished by higher caloric consumption, or that the benefits require treatment for longer than 28 days to become apparent. It might therefore be anticipated that a more prolonged exposure is required before any meaningful effect is apparent.
In conclusion, SRT2104 appears to be well tolerated in patients with T2DM but has no demonstrable beneficial effects on a range of measures of cardiovascular health. It is possible that while short-term exposure to SRT2104 is effective in mediating weight loss, it appears to be associated with an inadequate effect on diminishing insulin resistance, thereby causing deterioration in glycaemic control. Further larger-scale studies are required to confirm or refute these preliminary findings.
Acknowledgments
The authors thank the staff of the Wellcome Trust Clinical Research Facility in Edinburgh for their help with this study. The authors also thank Alison Hinds and Michelle Rostant-Belle from the Scottish Primary Care Research Network for their help with recruitment and the colleagues at Sirtris Pharmaceuticals Inc, Cambridge, Massachusetts, for their support throughout the study.
References
Footnotes
Contributors DEN is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis. RMN and SV were the main researchers who recruited the volunteers and conducted the study. JL, SD and NLM had input in writing the study protocol. RMN prepared the main manuscript and all other authors reviewed and edited the contents.
Funding The study was funded and supported by Sirtris Pharmaceuticals Inc, Cambridge, Massachusetts. They also supplied the study drug SRT2104 and its matching placebo.
Disclaimer DEN has undertaken consultancy for GSK; SD is currently an employee ofGlaxoSmithKline, UK, and owns stock; EH is an employee of Sirtris Pharmaceuticals, Massachusetts, and owns stock; BW is an employee of GlaxoSmithKline, Pennsylvania, and owns stock. RMN, SV, NLM, NNL and BMF declare no conflicts of interest.
Competing interests None declared.
Ethics approval Lothian Regional Ethics Committee.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement All available data can be obtained by contacting the corresponding author.